

Heloisa Moraes do Nascimento
DOI: 10.17545/e-oftalmo.cbo/2015.43
RESUMO
Uveíte significa inflamação da úvea, camada vascular do olho. Suas causas podem ser infecciosas, autoimunes ou secundárias. O diagnóstico e tratamento dependem do local da úvea acometido primariamente e causa de base. Este artigo fala sobre os diferentes tipos de uveíte e suas particularidades.
Palavras-chave: Uveíte, Doenças da Úvea, Toxoplasmose, Corticosteroides, Retina, Terapêutica
ABSTRACT
Inflammation of the uvea, which is the vascular layer of the eye, is termed uveítis. Uveítis may be caused by infections, autoimmune disorders, or secondary conditions. The diagnosis and treatment of uveítis depend on the part of the uvea that is primarily affected and the underlying cause. This article describes the different types of uveítis and their specificities.
Keywords: Uveitis, Uveal Deseases, Toxoplasmosis, Adrenal Cortex Hormones, Retina, Therapeutics
INTRODUÇÃO
O termo uveíte significa inflamação da úvea (íris, corpo ciliar e coroide). Sua importância advém do fato de que é responsável por 10% de pacientes com cegueira legal no mundo e pode acometer pacientes de qualquer idade. No Brasil, é responsável por 15,7% dos pacientes que freqüentam instituições de reabilitação visual. As possíveis causas são doenças infecciosas, inflamatorias ou secundárias como trauma, neoplasias, etc.
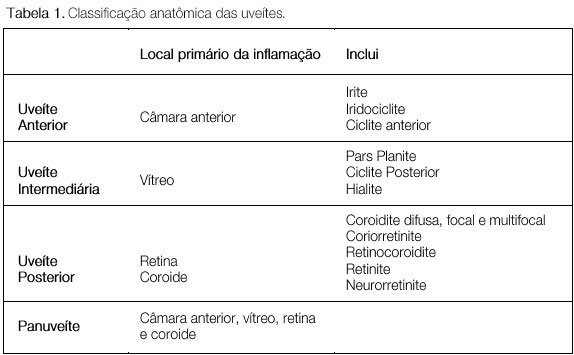
CLASSIFICAÇÃO DAS UVEÍTES
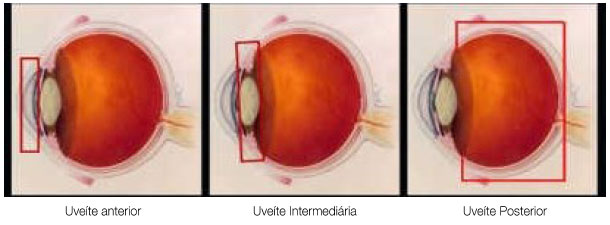
A classificação das uveítes auxilia o diagnóstico diferencial e a conduta. De acordo com os critérios do SUN (Standardization Uveitis Nomenclature)1, envolve a identificação do sítio primário de inflamação, tempo de aparecimento da doença e característica da inflamação (Tabelas 1 e 2 e Figura 1).

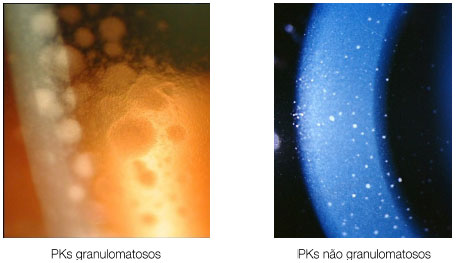
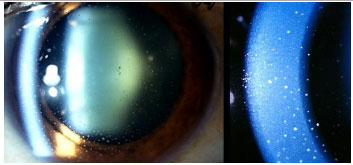
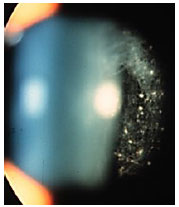
Os precipitados ceráticos (PKs) são aglomerados celulares localizados na superfície posterior da córnea e devem ser examinados cuidadosamente. Quando são grandes (mutton-fat), são conhecidos como granulomatosos e quando são pequenos e finos são chamados não granulomatosos. A coloração acinzentada com uma rede de fibrina entre eles sugere ser recente, enquanto a coloração pigmentada sugere ser mais antigo. Geralmente os PKs ficam localizados no triângulo de Arlt, na porção mais inferior da córnea. Entretanto, algumas uveítes podem apresentar PKs dispersos em toda a superfície posterior da córnea (Fuchs, Sarcoidose) enquanto que no Herpes tendem a se dispor na região corneana acometida (Figura 2).
A atividade da inflamação ocular pode ser mensurada de acordo com a quantidade de células presentes na câmara anterior. Para quantificá-la, a luz da lâmpada de fenda é regulada em sua intensidade máxima, usando-se a fenda de 1x1 mm e direcionando-se o raio de luz em um ângulo oblíquo ao plano iriano de 45-60 graus (Tabela 3 e Figura 3 e 4).



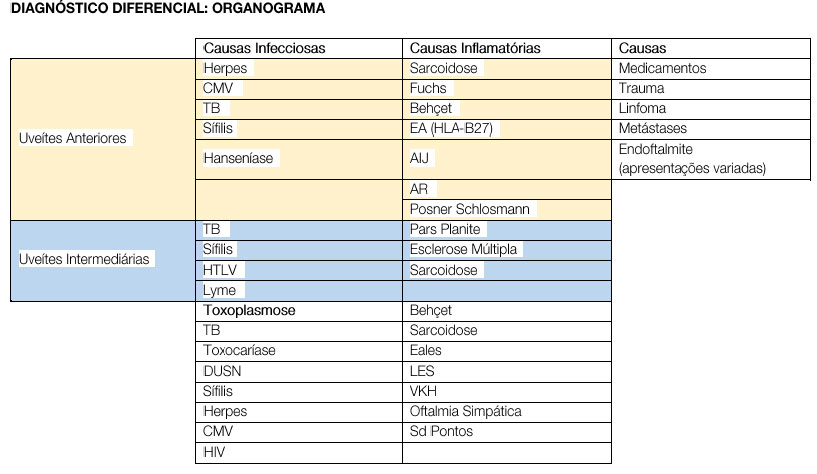
DIAGNÓSTICO DIFERENCIAL
Os sinais e sintomas variam de acordo com a causa específica da uveíte e o sítio primário de inflamação. As uveítes anteriores agudas frequentemente causam dor, fotofobia, hiperemia ocular e visão embaçada, entretanto pacientes com uveítes anteriores crônicas podem referir apenas leve diminuição da acuidade visual às vezes secundária a complicações como edema macular, catarata ou ceratopatia em faixa. A queixa principal dos pacientes nas uveítes intermediárias são opacidades no campo visual (floaters) e grandes moscas volantes eventualmente com baixa acuidade visual. Pacientes com uveítes posteriores frequentemente queixam-se de perda de visão, moscas volantes, fotopsias, metamorfopsias, escotomas, nictalopia ou uma combinação das queixas anteriores.
No Brasil, as causas infecciosas são responsáveis pela maioria dos casos de uveítes e devem ser sempre consideradas primariamente. Após sua exclusão, as causas inflamatórias devem ser pesquisadas. Doenças como artrite reumatoide, espondilite anquilosante, artrite idiopática juvenil, lúpus eritematoso sistêmico, doença de Behçet e sarcoidose podem apresentar a uveíte como primeira e única forma de manifestação ocular. Raramente, causas secundárias, como trauma, síndromes mascaradas como linfoma intraocular ou metástases podem causar quadros sugestivos de uveíte.
CICLITE HETEROCRÔMICA DE FUCHS
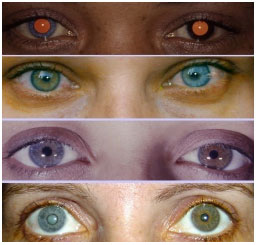
Uveíte anterior não granulomatosa, unilateral em 90% dos casos, de início insidioso e caráter crônico que acomete igualmente homens e mulheres. A tríade da doença é composta por uveíte + heterocromia + catarata. Com relação aos sinais caraterísticos da doença, geralmente o olho mais claro é o acometido, entretanto em olhos escuros a heterocromia às vezes é difícil de ser percebida, e em olhos azuis muito claros o olho mais escuro é o acometido. Os sintomas são leves, não há hiperemia importante e os PKs são brancos, difusos, estrelados, nunca confluentes. A catarata mais frequentemente associada é a subcapsular posterior. Uma hemorragia filiforme no ângulo após paracentese de câmara anterior pode acontecer (sinal de Amsler).
Glaucoma pode estar associado em até 20% dos casos. O tratamento deve ser individualizado de acordo com a inflamação ocular e muitas vezes é quase desnecessário. Midriáticos não são necessários porque não é uma uveíte sinequiante. O excesso de tratamento com corticoïdes tópicos ou sistêmicos pode levar a iatrogenias como aumento da pressão intraocular e progressão da catarata (Figura 5 e 6).
Síndrome de Posner Schlosmann: Uveíte anterior não granulomatosa, recorrente que acomete principalmente adultos jovens do sexo masculino. Classicamente apresenta-se como urna irite leve a moderada associada a um aumento intenso da pressão intraocular unilateral, que pode chegar a 60 mmHg. O ángulo da cámara anterior é aberto e não apresenta sinéquias posteriores. Entre as crises, o exame oftalmológico pode ser normal, à exceção de eventual atrofia óptica glaucomatosa. Durante as crises, o tratamento é realizado com esferoides tópicos e medicamentos antiglaucomatosos. Na maioria dos pacientes o prognóstico é bom, mas 25% dos casos podem evoluir para glaucoma.
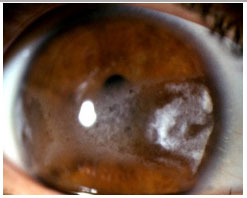
Uveíte anterior associada ao HLA-B27: Uveíte anterior não granulomatosa, sinequiante bilateral e assimétrica que acomete principalmente homens jovens, acometendo alternadamente os olhos. Mais de 50% dos pacientes com uveíte relacionada ao HLA-B27 tem uma doença sistêmica associada como espondilite anquilosante, síndrome de Reiter, doença inflamatória intestinal ou artrite psoriásica. Além dos sintomas oculares, os pacientes podem queixar-se de dor lombar, artrite, psoríase, úlceras orais, diarreia crónica e uretrite. A uveíte é aguda e recorrente, mas pode ser crônica. Os sintomas na fase aguda são intensos, com muita hiperemia conjuntival, fotofobia e dor. Sem tratamento, a crise demora semanas a meses, costumando recorrer. Urna intensa reação de cámara anterior com hipópio geralmente fibrinoso pode ser observada, assim como sinéquias posteriores. O tratamento inclui corticoïdes tópicos ou perioculares, midriáticos-cicloplégicos e corticoïdes sistêmicos (Figura 7).
Artrite idiopática juvenil: Uveíte anterior bilateral, não granulomatosa, sinequiante, crônica, geralmente assintomática que acomete principalmente meninas com artrite idiopática juvenil oligoarticular. Pode não ser percebida pela família, pois os olhos não ficam inflamados externamente e não há sintomas dolorosos. Muitas vezes é diagnosticada somente quando há complicações como catarata, ceratopatia em faixa e glaucoma. O tratamento inclui corticoïdes e midriáticos tópicos além de corticoïdes sistêmicos e imunossupressores. Não há nenhum biológico com indicação formal para a uveíte, mas em casos mais graves são frequentemente tentados com resultados muito variáveis e não comprovadamente superiores aos esferoides em longo prazo. A baixa visual é geralmente associada à ambliopia, catarata e ceratopatia em faixa. O implante de lente intraocular está relacionado a maior frequência de complicações como formação de membrana pupilar e opacidades retrolenticulares, e por esse motivo muitas vezes é contraindicado (Figura 8).
Artrite reumatoide (AR): Uveíte anterior não granulomatosa, bilateral que acomete geralmente mulheres de meia idade. As diversas manifestações oculares incluem: esclerite, ceratite ulcerativa periférica e uveíte anterior. O tratamento é baseado em esteroides tópicos ou perioculares e midriáticos. A doença sistêmica deve ser sempre tratada de acordo com sua atividade.
Uveítes traumáticas: A gravidade depende do tipo e da intensidade do trauma, mas classicamente é uma uveíte aguda, limitada, não granulomatosa, de intensidade leve a moderada, que responde bem a tratamento com corticoïdes tópicos. Midriáticos tópicos e analgésicos podem ser necessários. Dependendo do tipo do trauma, a presença de corpo estranho intraocular deve ser sempre suspeitada principalmente em uveítes crônicas unilaterais.
Herpes simples: Uveíte anterior granulomatosa ou não granulomatosa, geralmente unilateral, recorrente, que se caracteriza por edema de córnea com PKs atrás do edema e transiluminação setorial de íris. Associada ou não à ceratite e aumento da pressão intraocular, é muito mais sintomática e grave em crianças. Muitas vezes há desenvolvimento de neovasos estromais cornéanos profundos. O tratamento envolve antivirais sistêmicos e cicloplégicos-midriáticos. O vírus do Herpes também pode causar uma uveíte posterior que será descrita posteriormente (Figura 9).
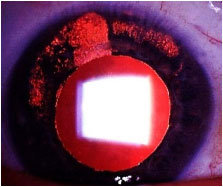
CMV: Muito raramente o vírus do CMV pode causar uveíte anterior, unilateral, não granulomatosa, recorrente e hipertensiva. Os PKs muitas vezes apresentam forma ameboide ou numular. Deve ser tratada com antivirais, corticoïdes e midriáticos tópicos (Figura 10).
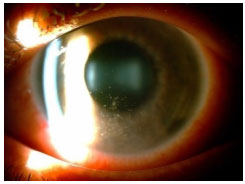
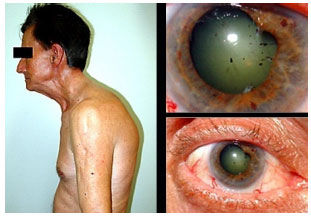
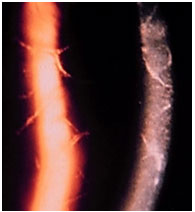
Hanseníase: Doença granulomatosa, crónica, causada pelo Mycobacterium leprae. O acometimento ocular é mais freqüente na hanseníase virchowiana. Pode cursar com madarose, diminuição da sensibilidade corneana, espessamento dos nervos cornéanos, uveíte anterior, intermediária e posterior. As pérolas irianas são sinais característicos da doença. O tratamento sistêmico envolve rifampicina, clofazimina e dapsona de acordo com o estágio clínico da doença. Se houver uveíte, esta deve ser tratada com esteroides e midriáticos tópicos (Figura 11 e 12).
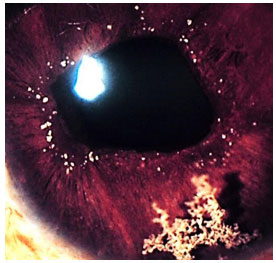
Sífilis e Tuberculose serão discutidas mais adiante por serem possíveis causas de todos os tipos de uveítes: anterior, intermediária e posterior.
UVEÍTES INTERMEDIÁRIAS
As uveítes intermediárias são classicamente descritas como aquelas que têm o vítreo como sítio primário de inflamação, mas com frequência apresentam muitas alterações vasculares e de polo posterior com edema macular.
UVEÍTES INTERMEDIÁRIAS RELACIONADAS A CAUSAS NÃO INFECCIOSAS
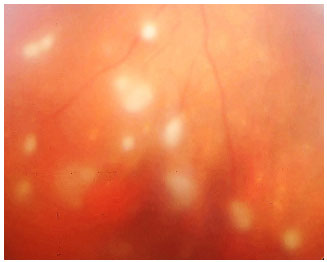
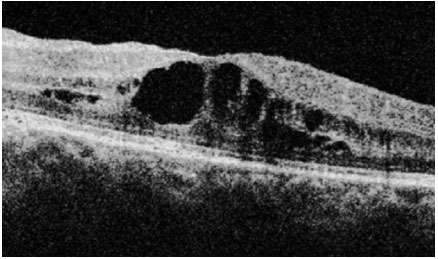
Pars Planite: Toda uveíte intermediária idiopática recebe o nome de pars planite. É o tipo de uveíte mais relacionado ao edema macular cistoide. A inflamação é muitas vezes assintomática e pode ser percebida apenas quando há complicações relacionadas à inflamação. A sintomatologia principal é a de opacidades flutuantes no campo visual. Classicamente é uma doença crônica, mais freqüente em mulheres jovens. Os sinais incluem células vítreas, snowballs, condensações vítreas arredondadas que se situam principalmente no vítreo inferior e snowbanking, condensação vítrea maciça em forma de bancada na periferia inferior retiniana. O tratamento nas formas leves é de apenas observação. Quando a visão está comprometida, envolve esferoides perioculares ou intraoculares. Muitas vezes são necessários imunossupressores (Figura 13-16).

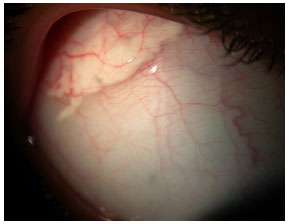
Sarcoidose: Doença granulomatosa sistêmica, crônica, de etiología desconhecida, menos freqüente em indivíduos da raça negra e mais em crianças e idosos. O acometimento sistêmico é principalmente pulmonar, e em até 50% dos casos podem ter comprometimento ocular. Geralmente estes pacientes apresentam anergia ao teste de PPD e tem história prévia de eritema nodoso, rash cutâneo, etc. A doença pode acometer qualquer parte do olho, incluindo pálpebras e glândula lacrimal. A forma mais comum de acometimento ocular é a uveíte granulomatosa bilateral, podendo esta ser anterior, intermediária, posterior ou pan-uveíte. O diagnóstico presumido de sarcoidose ocular é feito quando não há sinais de doença extraocular e o definitivo é realizado mediante a demonstração de granulomas não caseosos nos tecidos biopsiados. Os esferoides são a base do tratamento (Figura 17).
ESCLEROSE MÚLTIPLA
Doença desmielinizante que acomete principalmente mulheres jovens. Apresenta uveíte intermediária em 30% dos casos, sendo que ¼ desses pacientes apresentam uveíte antes que a doença desmielinizante seja diagnosticada. A uveíte tende a ser mais leve do que a observada em casos de pars planite. Deve-se estar atento a este diagnostico já que a esclerose múltipla é uma contraindicação ao uso de agentes biológicos, que muitas vezes são prescritos para pacientes com uveítes intermediárias de difícil controle.
UVEÍTES INTERMEDIÁRIAS RELACIONADAS A CAUSAS INFECCIOSAS
HTLV: O vírus linfotrópico humano de células T tipo I é um retrovirus de transmissão transplacentária, sexual, por transfusão sanguínea ou através de aleitamento materno, que é endêmico no Japão e no Brasil, com maior frequência na região Nordeste. Está relacionado à paralisia espástica, linfoma, leucemia e uveíte intermediária, crónica-recidivante e vasculite retiniana. O tratamento é feito com esteroides.
Doença de Lyme: Não há relatos de doença de Lyme no Brasil. Doença causada pela espiroqueta Borrelia burgdorferi endêmica nos Estados Unidos e alguns países da Europa e transmitida por picada de carrapatos. Causa eritema migratório, artrite, e raramente meningite. As manifestações oculares podem ser: conjuntivite folicular, uveíte anterior, intermediária e posterior, sendo a intermediária a forma mais comum de apresentação. O envolvimento ocular deve ser interpretado como comprometimento do sistema nervoso central e o tratamento envolve antibioticoterapia sistêmica.
UVEÍTES POSTERIORES
Uveítes posteriores relacionadas a causas não infecciosas
Síndrome de Pontos Brancos
Coriorretinopatia de Birdshot: Uveíte posterior crônica, rara, com vitreíte significativa, sem snowballs ou snowbankings, com exacerbações e remissões. Acomete mais mulheres brancas de meia idade. Predisposição genética evidenciada pela associação com HLA-A29. Pode haver edema macular. Na AF apresenta hipofluorescência precoce com hiperfluorescência tardia. O ERG é utilizado no monitoramento da doença (onda a preservada, redução da amplitude e aumento do tempo de latência da onda b). O EOG pode ser subnormal. O tratamento envolve esteroides locais e sistêmicos. O uso de agentes imunossupressores é muitas vezes necessário (Figura
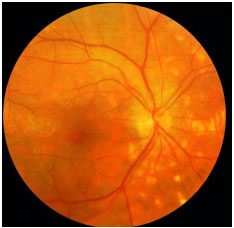
Coroidite Serpiginosa: Uveíte posterior rara bilateral crônica progressiva recorrente, com meses ou anos de inatividade entre os episódios, de etiología desconhecida. Acomete mais homens de meia idade. É geralmente assintomática até que a mácula seja acometida. O olho é calmo, com nenhuma ou mínima reação de câmara anterior e o vítreo pode apresentar finas células pigmentadas. As lesões ativas apresentam-se como infiltrados subretinianos acinzentados ou amarelados, que se iniciam junto ao disco óptico, com progressão centrífuga e irregular. A neovascularização da coroide é a complicação mais comum. Deve ser diferenciada da tuberculose ocular. AF: hipofluorescência precoce com margens hiperfluorescentes que aumentam em direção ao centro da lesão com extravasamento progressivo na fase tardia. O tratamento envolve esteroides locais e sistêmicos. O uso de agentes imunossupressores é muitas vezes necessário (Figura 19).
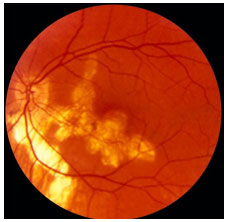
Síndrome dos múltiplos pontos brancos evanescentes (MEWDS): Distúrbio unilateral transitorio que acomete preferencialmente mulheres jovens, que apresentam múltiplos pontos brancos profundos na retina externa ou EPR, com aparência granular na mácula. Em cerca de 33 a 50% dos casos há relato de pródromo viral. Ocorrem BAV súbita, escotomas paracentrais e DPAR pode estar presente. A reação de câmara anterior é mínima ou ausente e a vitreíte é moderada. Pode haver hiperemia ou edema do disco óptico. Apresenta um curso agudo com média de 7 semanas e melhora espontânea. A perda de campo visual pode persistir. AF: Hiperfluorescência precoce com impregnação tardia. ERG: diminuição da onda a. O EOG também pode estar alterado (Figura 20).
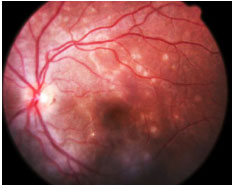
Epiteliopatia Pigmentária Placoide Multifocal Posterior Aguda (EPPMPA): Doença aguda com múltiplas lesões em placa branco-amareladas no polo posterior, com rápida perda visual e resolução espontânea em poucas semanas, deixando discretas alterações cicatriciais. Afeta igualmente homens e mulheres jovens. Geralmente bilateral. Um terço dos indivíduos apresenta pródromos virais. Tem associações com vasculites sistêmicas. AF: hipofluorescência precoce com hiperfluorescência tardia (Figura 21).
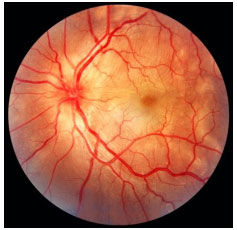
Coroidite multifocal e pan-uveíte: Uveíte crônica bilateral assimétrica caracterizada por múltiplas lesões coriorretinianas similares às observadas na síndrome da histoplasmose ocular presumida. Afeta principalmente mulheres idosas. Há reação de câmara anterior leve a moderada com precipitados ceráticos não granulomatosos e sinéquias posteriores. A vitreíte é moderada a grave. À fundoscopia, apresenta lesões branco amareladas de 50 a 100µm de diâmetro que primariamente envolvem a coroide e a retina externa. A causa mais freqüente de perda visual é a neovascularização coroideana. AF: hipofluor precoce com hiper tardia (Figura 22).
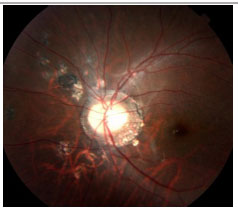
Retinopatia externa oculta zonal aguda (AZOOR): Perda aguda de uma ou mais zonas da retina externa associada com fotopsia, alterações fundoscópicas mínimas e achados anormais no ERG. Uni ou bilateral. Acomete mais frequentemente mulheres jovens míopes. Pode haver vitreíte leve e perda de campo visual. AF pode ser normal com apenas atraso do enchimento vascular. Não há consenso sobre o tratamento (Figura 23).
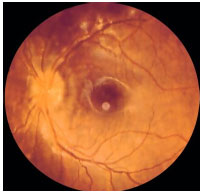
Coroidite Puntata Interna: Doença inflamatoria idiopática que acomete mulheres jovens míopes com queixa de metamorfopsia, escotoma paracentral, fotopsia e baixa acuidade visual. As lesões são pequenas (100 a 200 µm) e acometem o polo posterior. Não há vitreíte. Progridem para cicatrizes atróficas e podem apresentar membranas neovasculares subretinianas como complicação. Na AF, há hiperfluorescência precoce com hiperfluorescência tardia (Figura 24).
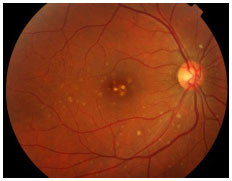
UVEÍTES POSTERIORES RELACIONADAS A CAUSAS INFECCIOSAS
Toxoplasmose
Causada pelo Toxoplasma gondii, protozoário adquirido pela ingestão de carne de porco ou frango mal passada, águas e alimentos contaminados por fezes de gato ou via transplacentária, é a principal causa de uveíte posterior no Brasil e no mundo. A infecção sistêmica é percebida como uma gripe e muitas vezes é assintomática.
A toxoplasmose ocular congênita ocorre quando a mãe adquire a infecção durante a gravidez. Quanto mais precoce no curso da gestação, mais grave a infecção fetal. No primeiro trimestre muitas vezes leva a abortamento. No acometimento ocular CLÁSSICO o recém-nascido apresenta lesões de retinocoroidite macular em roda de carroça. A Toxoplasmose congênita pode afetar os olhos muitos anos depois do nascimento (Figura 25).
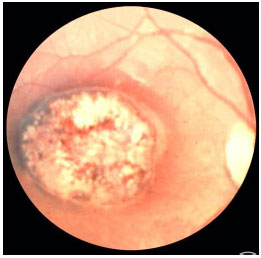
A doença é grave e por vezes leva a baixa visual permanente e cegueira legal. O tratamento da infecção no RN é geralmente durante 12 meses e deve ser feito em conjunto com o pediatra. Posteriormente a este período e dependendo da gravidade, as crianças devem receber profilaxia com Sulfametoxazol 800 mg + Trimetoprima 160 mg por vários anos para evitar crise subclínica uma vez que a criança não verbaliza os sintomas. A principal forma de evitar a toxoplasmose ocular congênita é educar as mães sobre a forma de contágio e orientações em relação à alimentação.
A toxoplasmose ocular adquirida manifesta-se como uma uveíte posterior granulomatosa com retinocoroidite necrosante exsudativa, classicamente adjacente a uma cicatriz de toxoplasmose prévia, com vitreíte localizada principalmente acima da lesão (sinal do farol na neblina). Os sintomas visuais dependem da localização da lesão na retina, mas em geral os pacientes queixam-se de moscas volantes, seguido por baixa acuidade visual (Figura 26 e 27).
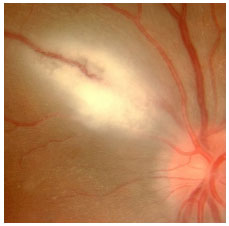
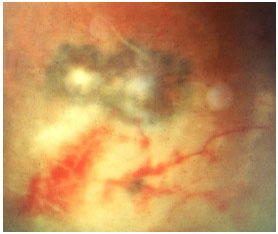
O tratamento clássico envolve o uso de sulfadiazina 4g/dia, pirimetamina 25mg/d, e corticoïdes quando necessário. O tratamento também pode ser feito com a combinação de sulfametoxazol 800mg + trimetropim 160mg. Pacientes alérgicos à sulfa podem usar clindamicina (300 mg VO 6/6 horas) como alternativa terapêutica. A injeção intravítrea de clindamicina e dexametasona também é considerada eficaz.
Pacientes com lesões oculares de toxoplasmose ocular graves e recidivas freqüentes podem beneficiar-se da profilaxia com sulfametoxazol+trimetropim três vezes por semana durante anos.
TUBERCULOSE
A tuberculose (TB), endêmica no Brasil, é uma doença granulomatosa sistêmica causada pelo Mycobacterium tuberculosis. Sua principal forma de apresentação é a tuberculose pulmonar. Entretanto, pode disseminar-se via hematogênica e causar infecção em outros órgãos.
As manifestações oculares são diversas, e podem envolver pálpebra, conjuntiva, íris, retina e até o nervo óptico. Não se sabe ao certo se as manifestações oculares são causadas pela própria micobactéria ou pelo estímulo imunogênico causado pela mesma. Classicamente pode apresentar-se como uma uveíte anterior granulomatosa, intermediária ou posterior (forma mais comum).
O teste PPD (purified protein derivative) é um teste de triagem que avalia exposição prévia á micobactéria e pode auxiliar no diagnóstico de TB ocular. Apesar de apresentar muitos resultados falso positivo, deve ser interpretado da seguinte maneira:
PPD anérgico (0 mm): paciente não vacinado, vacinado há muito tempo ou sugestivo de sarcoidose;
PPD 0-5mm: paciente vacinado recentemente;
PPD 5-1 Omm: deve ser considerado positivo em pacientes imunocomprometidos, com HIV, pacientes em contato com tuberculose ativa ou raio X de tórax consistente com lesões de tuberculose;
PPD 10-15mm: considerado positivo em pacientes diabéticos, com falência renal, em uso de imunossupressores, profissionais de saúde ou imigrantes de regiões de alta prevalência;
PPD> 15mm: considerado positivo em pacientes sem fatores de risco (Figura 28).
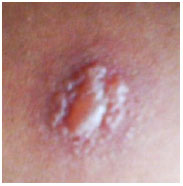
Novos testes baseados em interferon de linfócitos de sangue periférico apresentam maior especificidade e sensibilidade que o PPD e menor reatividade cruzada com a vacinação BCG e também podem auxiliar o diagnóstico de tuberculose. Entretanto, ainda apresentam custo elevado e restrições de realização no Brasil (Figuras 29-31).
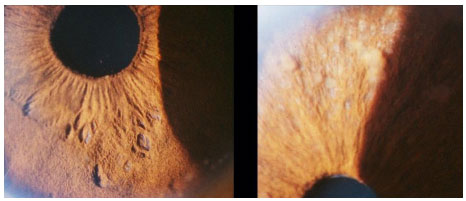
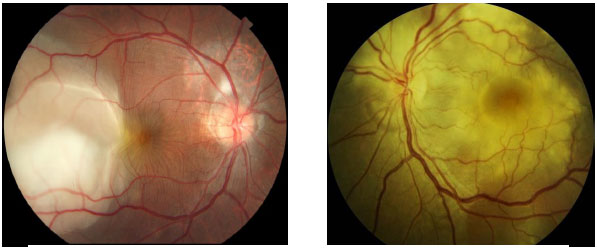
O tratamento da tuberculose ocular deve ser feito com o esquema quádruplo de rifampicina + izoniazida + pirazinamida + etambutol. O etambutol deve ser descontinuado caso haja toxicidade de nervo óptico. O Ministério da Saúde recomenda tratamento por seis meses. Entretanto, muitas vezes este deve ser prolongado em casos de TB ocular até por 15 meses. O uso de corticoïdes auxilia a melhora das manifestações oculares.
SÍFILIS
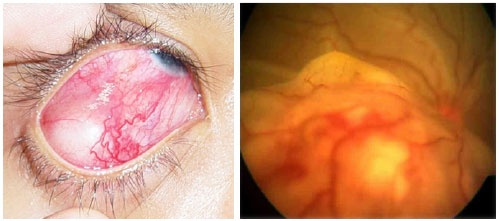
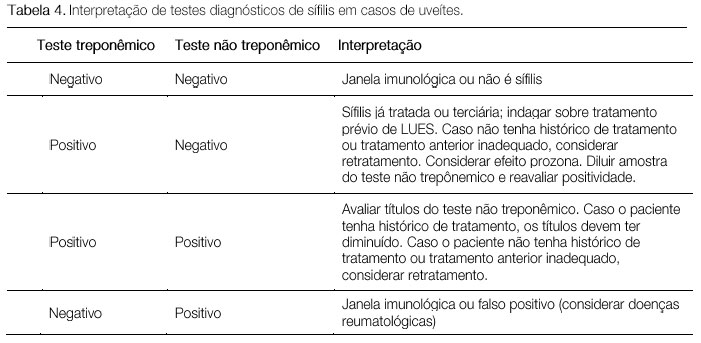
A sífilis é a grande mimetizadora entre as doenças inflamatorias oculares. Seu espectro clínico é muito variável e pode causar todos os tipos de uveíte. Portanto, deve ser sempre excluída em casos de inflamação ocular. A sífilis atinge o olho em seus estágios secundário e terciário. O modo de transmissão é através do contágio sexual, contato com sangue ou via transplacentária. O diagnóstico baseia-se em testes treponêmicos, tais como FTA-Abs e ELISA, e testes não treponêmicos, como o VDRL (venereal disease reseach laboratory). (Tabela 4) O tratamento, igual ao da neurossífilis, deve ser feito com penicilina cristalina endovenosa 4 milhões de Ul a cada 4 horas durante 14 dias. A resposta ao tratamento costuma ser muito boa.
Entretanto, pode ocorrer intensificação dos sintomas da doença após a administração do antibiótico, quadro conhecido como reação de Jarisch-Herxheimer. Clinicamente há piora nas lesões de pele, dor muscular, febre com calafrios e cefaleia. Sua ocorrência está relacionada a uma reação autoimune do paciente em resposta à liberação de toxinas pela bactéria após o tratamento. O uso de esteroides sistêmicos pode prevenir esta reação, além de acelerar a recuperação dos sinais e sintomas da uveíte (Figura 32).
UVEÍTES POSTERIORES VIRAIS
Necrose aguda de retina (ARN)
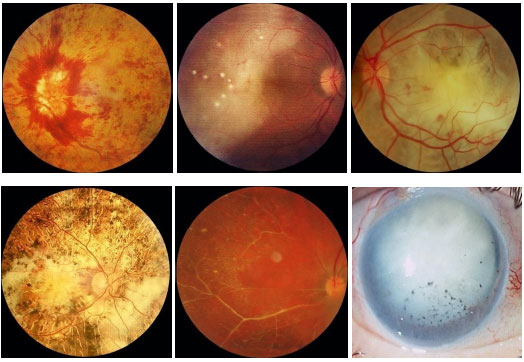
Entidade rara caracterizada por necrose aguda de toda a espessura da retina, causada pelo virus varicela zoster, herpes simples tipo 1 ou 2 e, raramente, CMV e EBV. Apesar de classicamente ser considerada uma doença de indivíduos jovens imunocompetentes, estudos recentes apontam que há uma predisposição em usuários crônicos de corticoïdes e em pacientes com imunossupressão relativa. O quadro clínico é caracterizado por uma uveíte anterior leve a moderada com aumento de pressão intraocular e a tríade de envolvimento do segmento posterior é composta por vitreíte moderada a severa, vasculite oclusiva e retinite nécrosante que inicialmente acomete a periferia da retina e apresenta rápida progressão circunferencial. O segundo olho é acometido em um terço dos casos em 1 a 6 semanas; entretanto, há relatos com intervalos maiores de 30 anos. O tratamento deve ser feito prontamente com aciclovir endovenoso (10-15 mg/kg 8/8 horas por 15 dias). O cloridrato de Valaciclovir também pode ser usado pela vantagem de via oral e boa biodisponibilidade, mas não há estudos comparativos. Com tratamento, o quadro geralmente se resolve em 3 semanas. Sem tratamento, o curso da doença é mais prolongado (cerca de 12 semanas). O tratamento reduz a ocorrência de doença no olho contralateral, mas persiste o risco mesmo em períodos de muitos anos. O afinamento da retina acometida associado à infiltração celular vítrea predispõe ao desenvolvimento de buracos e roturas retinianas que, por sua vez, levam ao descolamento de retina regmatogênico em 75% dos casos. Não há consenso sobre a vitrectomia precoce e o uso de terapia a laser no curso da necrose aguda de retina (Figura 33).
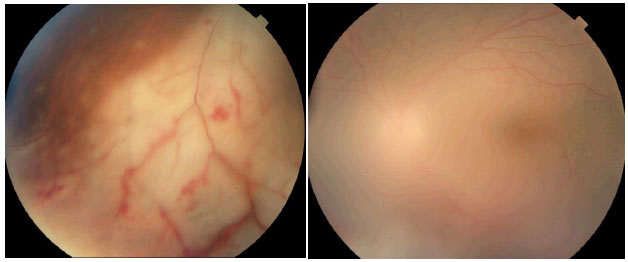
NECROSE PROGRESSIVA DA RETINA EXTERNA (PORN)
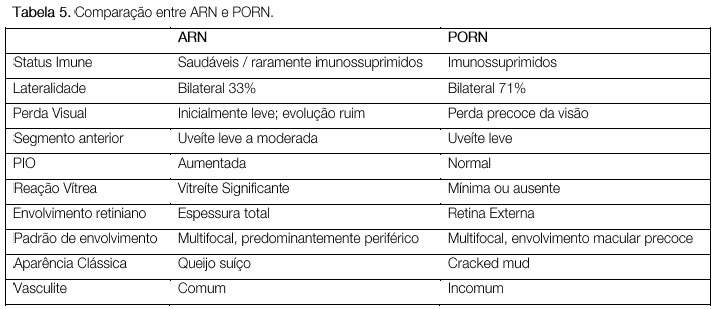
PORN (progressive outer retinal necrosis) é uma entidade rara, descrita em 1990. Assim como a ARN, é causada pelo vírus herpes simples tipo 1 e 2 ou varicela zoster, entretanto é mais frequentemente observada em indivíduos HIV positivos com grau severo de imunossupressão (CD4<50). Característicamente há uma necrose aguda de retina externa, rapidamente progressiva, bilateral em cerca de 2/3 dos casos, com lesões multifocais que acometem inicialmente o polo posterior. O prognóstico é muito ruim e o tratamento é o mesmo da necrose aguda de retina (Tabela 5 e Figura 34).
CITOMEGALOVIRUS
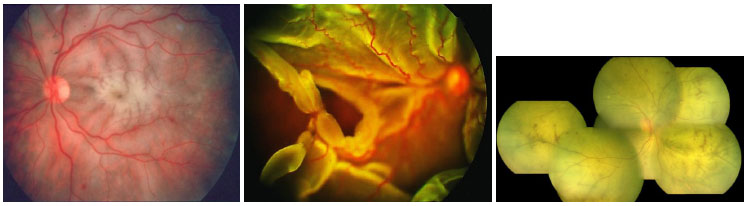
A retinite causada pelo citomegalovírus (CMV) é a principal doença ocular relacionada ao HIV. Na era pré-HAART, era observada em mais de 40% dos pacientes. Atualmente houve um declínio na incidência, variando de 5 a 10% em diferentes países. Os pacientes que desenvolvem retinite por CMV geralmente apresentam CD4 <50/µL (média de 17/µL) e queixam-se de baixa acuidade visual associada a moscas volantes e perda de campo visual, mas pode ser assintomática em 15% dos casos.
O quadro clínico é caracterizado por uma retinite necrosante não exsudativa que se inicia ao redor dos vasos e progride centrípetamente, geralmente associada a hemorragias (lesão em “queijo com catchup”). O comprometimento anterior é leve, com pouca ou nenhuma reação de câmara anterior e PKs finos no endotélio. A vitreíte também é leve.
As formas clínicas são: típica ou hemorrágica, atípica ou granular e retinite perivascular (frosted branch angiitis),esta última menos comum. As lesões cicatrizam deixando a retina atrófica e predispondo a descolamento de retina regmatogênico (Figura 35-37).
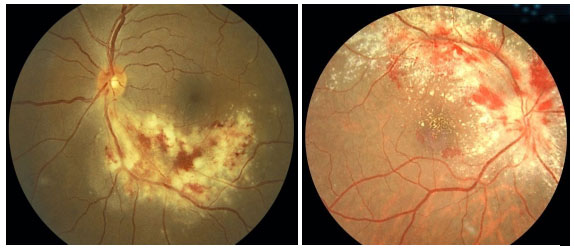
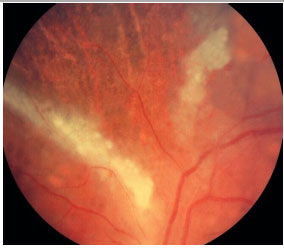
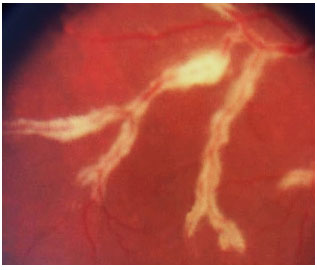
O tratamento padrão envolve agentes antivirais como ganciclovir, foscarnet, cidofovir endovenosos e valganciclovir oral. O tratamento de indução é feito por 2 a 4 semanas e o tratamento de manutenção deve ser continuado até que os níveis de CD4 estejam acima de 150/µL. A administração sistêmica desses medicamentos pode causar graves efeitos colaterais como neutropenia grave secundária ao ganciclovir e falência renal secundária ao tratamento com foscarnet e cidofovir. Implantes intraoculares de ganciclovir estão disponíveis em alguns países, mas não no Brasil. Laser pode ser feito nas áreas predispostas a descolamento. Se necessário, vitrectomia via pars plana associada a endolaser nas microroturas e colocação de óleo de silicone são indicadas. Os resultados com implante de óleo são melhores do que com gás (C3F8 ou SF6).
Desde a introdução do HAART, uma nova entidade denominada SÍNDROME DA RECUPERAÇÃO IMUNE surgiu. Trata-se de um distúrbio inflamatório crônico que ocorre com a melhora da imunidade geral do paciente e melhora dos níveis de CD4. O quadro clínico é caracterizado por intensa uveíte anterior e vitreíte que podem estar associados a papilite e edema macular cistoide. A manutenção da medicação anti CMV aumenta a incidência desta síndrome em especial nos casos de tratamento com cidofovir. O tratamento é baseado em corticoides.
PAN-UVEÍTES
Síndrome de Vogt-Koyanagi-Harada
A síndrome de Vogt Koyanagi Harada é uma doença rara de etiología desconhecida que envolve o olho, a meninge e o encéfalo (uveo-meningo-encefalite). Vários espectros da doença permitem que ela se manifeste como uma uveíte posterior (Doença de Harada) ou majoritariamente como pan-uveíte (Síndrome de Vogt Koyanagi Harada - VKH).
O quadro agudo envolve pan-uveíte granulomatosa com descolamento seroso de retina bilateral, associado a cefaleia e zumbido. O exame de liquor pode revelar pleocitose. Às vezes, o descolamento de retina pode ser muito extenso e confundir-se com um descolamento regmatogênico (Figura 38).
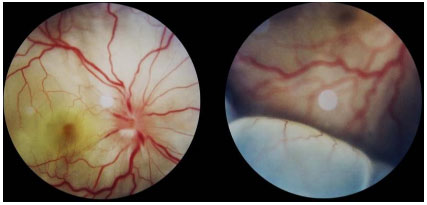
A doença aguda deve ser tratada agressivamente com pulsoterapia com metilprednisolona 1 g/d por 3 dias, associado à profilaxia para estrongiloides (Albendazol 400 mg VO por 3 dias). Após a pulsoterapia, o paciente deve ser mantido com esteroides orais, diminuindo a dose conforme a resposta do paciente. Pelo caráter severo e recidivante, todos os pacientes com VKH são candidatos à imunossupressão sistêmica prolongada ou crônica. Corticoïde intravítreo pode rapidamente diminuir a uveíte e o descolamento de retina, devendo, no entanto, ser sempre associado ao tratamento sistêmico.
As constantes recidivas manifestam-se como novos episódios de pan-uveíte granulomatosa associada a descolamento seroso de retina. O quadro crônico revela uma característica despigmentação fundoscópica e rearranjo do epitélio pigmentado da retina, conhecido como fundus em sunset glow. Também podem ser observadas pequenas manchas amareladas dispersas principalmente na periferia e média-periferia da retina conhecidas como nodulos de Dallen-Fuchs (Figura 39 e 40).
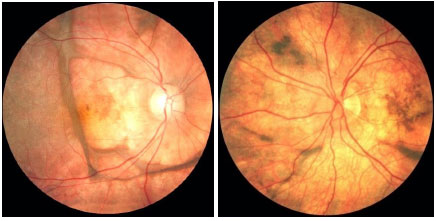
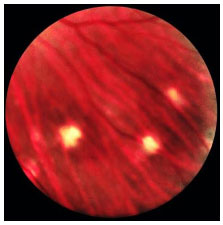
As manifestações tardias da doença incluem alterações cutâneas como vitiligo, poliose e alopecia (Figura 41).
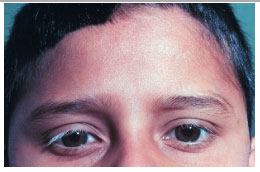
A síndrome de Vogt-Koyanagi-Harada é urna uveíte grave que pode levar à perda visual permanente. Portanto, o diagnóstico precoce e tratamento adequado são fundamentais para minimizar os danos que esta condição pode causar.
OFTALMIA SIMPÁTICA
A oftalmía simpática (OS) é uma entidade muito rara que acomete individuos com histórico de trauma ocular penetrante, na maioria das vezes com exposição de tecido uveal ou cirurgia intraocular prévia. O tempo entre o trauma e o aparecimento da inflamação é bastante variável e esta condição é muitas vezes subdiagnosticada. A maioria dos pacientes desenvolve uma pan-uveíte granulomatosa grave no olho contralateral (simpatizante). Os sintomas variam desde irritação ocular e moscas volantes até baixa de acuidade visual severa. Pode haver descolamento seroso de retina e, muitas vezes, a doença comporta-se de forma muito semelhante à síndrome de Vogt-Koyanagi-Harada, podendo levar à cegueira. Tardiamente no curso da doença, também é possível observar nodulos de dallen-fuchs no fundo do olho. O tratamento deve ser precoce e agressivo, envolve uso de esferoides na fase aguda e posteriormente imunossupressão sistêmica conforme necessário (Figura 42 e 43).
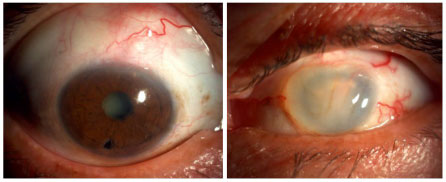
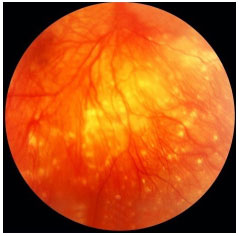
DOENÇA DE BEHÇET
A doença de Behçet é uma vasculite oclusiva não granulomatosa sistêmica crônica de etiología desconhecida, associada ao HLA-B51, mais freqüente na Ásia, oriente médio e também no Brasil. O diagnóstico é clínico e envolve a presença de úlceras orais dolorosas, úlceras genitais, uveíte, assim como alterações cutâneas (Tabela 6).
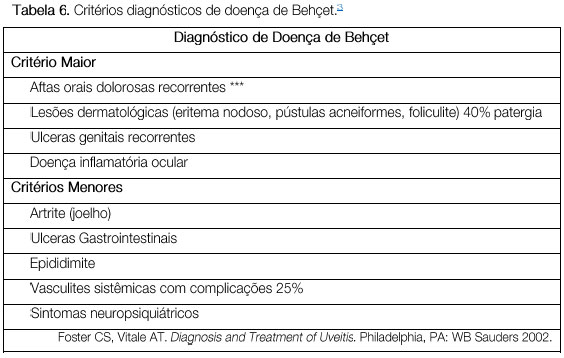
A doença é considerada completa quando apresenta 4 critérios maiores, incompleta quando apresenta 3 critérios maiores ou 1 critério maior associado a envolvimento ocular, suspeita quando apresenta 2 critérios maiores sem envolvimento ocular e possível quando apresenta 1 critério maior. A uveíte, observada em 70% dos pacientes com comprometimento sistêmico, pode ser o primeiro sintoma da doença. Geralmente é uma pan-uveíte bilateral com hipópio móvel e vasculite dos vasos retinianos. Se não tratada adequadamente e precocemente durante as crises, pode levar a perda visual permanente (Figura 44 e 45).
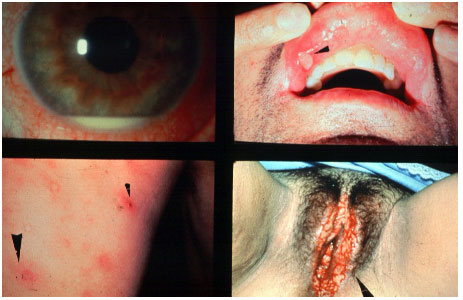
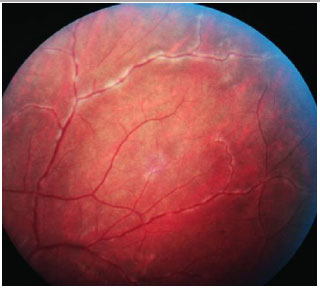
ENDOFTALMITE
Diagnóstico clinico caracterizado por intensa inflamação intraocular secundária a infecção bacteriana ou fúngica. Além do comprometimento dos segmentos anterior e posterior, podem ser observadas ceratite ou esclerite. Os sinais clássicos de endoftalmite são embasamento visual, dor, hiperemia e quemóse conjuntival, edema palpebral e hipópio.
As endoftalmites podem ser classificadas em exógenas (pós-cirúrgica aguda ou tardia e traumática) e endógenas (menos comuns). O diagnóstico de endoftalmite é clínico. Entretanto, exames complementares de imagem podem ser necessários, principalmente quando há suspeita de corpo estranho intraocular. Amostras de humor aquoso e vítreo devem ser sempre coletadas para confirmação do agente etiológico. O tratamento deve ser dirigido ao agente etiológico de base com antibiótico ou antifúngico tópico, intravítreo ou sistêmico, associado a corticoïdes tópicos e midriáticos. Qualquer atraso no tratamento pode piorar o prognostico visual e levar à cegueira.
ENDOFTALMITE AGUDA PÓS-CIRÚRGICA
Ocorre classicamente nas primeiras 6 semanas após cirurgia intraocular. A incidência varia entre 0,07%-0,12%. Os agentes etiológicos mais relacionados são os staphylococcus coagulase negativos, mas staphylococcus aureus, streptococcus sp e bactérias GRAM negativas também foram isoladas. Fatores de risco pré-operatórios conhecidos são imunossupressão sistêmica (diabetes, câncer), blefaroconjuntivites, etc. Entre os fatores de risco intraoperatórios podemos citar: rotura capsular, prolapso de íris, cirurgias prolongadas, perda vítrea, etc. Uma intensa reação de câmara anterior, com hipópio e vitreíte importante são frequentemente observados. O quadro deve ser diferenciado da Síndrome Tóxica do segmento Anterior (TASS) que ocorre mais precocemente (1-2 dias pós-op) e caracteriza-se por ausência de dor e comprometimento mínimo do segmento posterior.
ENDOFTALMITE CRÔNICA PÓS-CIRÚRGICA (> 6 SEMANAS ATÉ 2 ANOS APÓS A CIRURGIA INTRAOCULAR)
Os agentes etiológicos mais comumente associados são Propionibacterium acnes, fungos e bactérias Gram positivas e negativas menos virulentas. Os sinais e sintomas são menos severos e o início mais insidioso em relação às endoftalmites agudas pós-cirúrgicas. Um sinal característico pode ser a presença de uma placa esbranquiçada no saco capsular ou o aparecimento súbito de hipópio em um paciente com inflamação crônica pós-operatória.
ENDOFTALMITE TRAUMÁTICA
Este tipo de endoftalmite pode ocorrer depois de qualquer trauma ocular penetrante. Os agentes etiológicos mais associados são: S. aureus e epidermidis, Bacillus sp, Streptococcus sp e vários fungos. Um retardo > 12 horas na reparação cirúrgica aumenta o risco de endoftalmite e piora o prognóstico visual, especialmente nos pacientes com suspeita de corpo estranho intraocular.
ENDOFTALMITE ENDÓGENA
Inflamação intraocular secundária a disseminação hematôgena de uma sepse sistêmica. Uma boa história e um alto grau de suspeita clínica são fundamentais nesses casos, pois infeções como endocardites, e infeções de pele e trato urinário podem passar despercebidas. Fatores de risco associados são: imunossupressão sistêmica, uso de drogas ou corticoterapia crônica, cateteres intravenosos, alcoolismo, hepatopatias e cirurgias abdominais recentes. Na maioria dos casos, os sintomas sistêmicos são inespecíficos, e podem incluir febre, mialgias, etc. O comprometimento ocular é bilateral em até 25% dos casos e pode incluir: nódulos irianos ou microabscesos, reação de câmara anterior mínima ou severa (com hipópio inclusive), placas coroideas esbranquiçadas, com comprometimento retiniano progressivo, exsudados algodonosos, vasculite retiniana e vitreíte. O tratamento é direcionado à infeção de base, sendo a identificação do agente causal de suma importância para a seleção da melhor terapia antibiótica ou antifúngica (Figura 46).
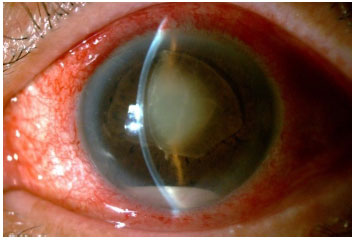
VASCULITE
As vasculites podem ser idiopáticas isoladas ou associadas a qualquer vasculite sistêmica, doenças infecciosas ou tumorais. O diagnóstico geralmente é clínico, mas a angiofluoresceinografia (AFG) é de fundamental importância para a documentação e seguimento do quadro clínico, pois geralmente o comprometimento vascular retiniano é mais extenso do que o exame fundoscópico sugere. O aspecto fundoscópico característico é de uma exsudação ao redor dos vasos ou de um embainhamento das paredes vasculares. Na AFG geralmente é observada uma hiperfluorescência perivascular focal (sarcoidose e esclerose múltipla) ou difusa (Behcet, Birdshot e doença de Eales).
As arterites são mais frequentemente observadas em Lúpus eritematoso sistêmico, poliarterite nodosa, IRVAN, síndrome de Churg Strauss, sífilis, HSV (ARN) e VZV (PORN). As flebites são clássicas da sarcoidose, esclerose múltipla, doença de Behcet e doença de Eales. A combinação das anteriores é geralmente vista em uveítes associadas à Toxoplasmose, Policondrite Recidivante, Granulomatose de Wegener, Doença de Crohn, etc. Quando a inflamação vascular não está associada a doenças infecciosas, o diagnóstico diferencial pode ser difícil e requerer uma ampla investigação sistêmica. O tratamento é sempre orientado à doença de base e muitas vezes é necessária uma imunossupressão prolongada e fotocoagulação com laser para evitar a progressão e possíveis complicações como hemorragia vítrea, descolamento de retina tracional e glaucoma neovascular.
REFERÊNCIAS
1 A A Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveítis Nomenclature (SUN) Working Group. Standardization of uveítis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005;140:509-516. http://dx.d0i.0rg/l 0.1016/i.aio.2005.03.057
2. Oréfice e Belfort. Atlas Latino-Americano de Uveítes. Aplicativo disponível para iPhone e Ipad em AppIeStore, 2013.
3. A Foster CS, Vitale AT. Diagnosis and Treatment of Uveítis. Philadelphia, PA: WB Sauders, 2002.
LEITURA COMPLEMENTAR:
Silveira C, Belfort R Jr, Muccioli C, et al. The effect of long-term intermitent trimethoprim/sulfamethoxazole treatment on recurrences of toxoplasmic retinochoroiditis. Am J Ophthalmol. 2002:134(11:41-46 http://dx.d0i.0rg/l 0.1016/S0002-9394(02)01527-1
Nussenblatt, Robert B. Uveítis: fundamentals and clinical practice. Pennsylvania: Mosby; 2004.
Intraocular Inflammation and Uveítis 2013-2014 - Basic and Clinical Science Course - Section 9 - American Academy of Ophthalmology.
Oréfice, Fernando, Uveíte - Clínica e Cirúrgica. Cultura Médica. Segunda Edição; 2005.

Fonte de financiamento: declaram não haver.
Conflito de interesses: declaram não haver.
Recebido em:
11 de Janeiro de 2016.
Aceito em:
26 de Fevereiro de 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket