

Heloisa Moraes do Nascimento
DOI: 10.17545/e-oftalmo.cbo/2015.43
ABSTRACT
Inflammation of the uvea, which is the vascular layer of the eye, is termed uveítis. Uveítis may be caused by infections, autoimmune disorders, or secondary conditions. The diagnosis and treatment of uveítis depend on the part of the uvea that is primarily affected and the underlying cause. This article describes the different types of uveítis and their specificities.
Keywords: Uveitis, Uveal Deseases, Toxoplasmosis, Adrenal Cortex Hormones, Retina, Therapeutics
RESUMO
Uveíte significa inflamação da úvea, camada vascular do olho. Suas causas podem ser infecciosas, autoimunes ou secundárias. O diagnóstico e tratamento dependem do local da úvea acometido primariamente e causa de base. Este artigo fala sobre os diferentes tipos de uveíte e suas particularidades.
Palavras-chave: Uveíte, Doenças da Úvea, Toxoplasmose, Corticosteroides, Retina, Terapêutica
INTRODUCTION
“Uveitis” means inflammation of the uvea (iris, ciliary body, and choroid). This condition is important because it accounts for 10% of patients with legal blindness worldwide and affects people of all ages. In Brazil, it accounts for 15.7% of patients who attend visual rehabilitation centers. Possible causes are infections, inflammatory diseases, and secondary to conditions such as trauma and tumors.
CLASSIFICATION OF UVEITIS
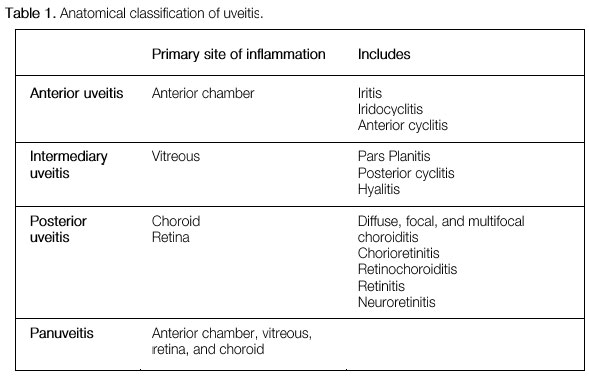
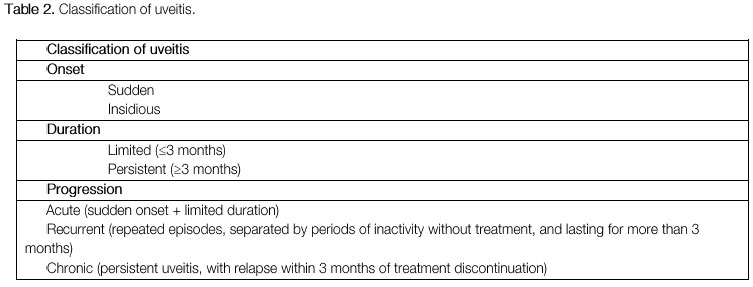
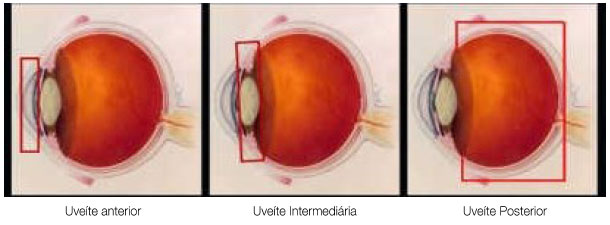
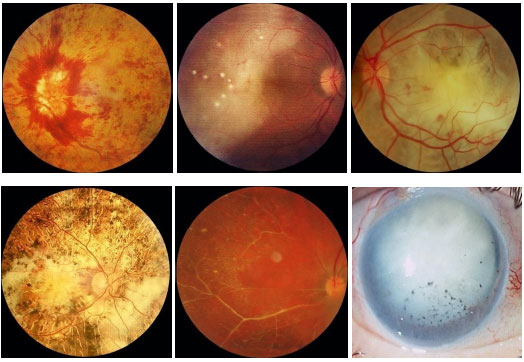
The classification of uveitis is useful for the differential diagnosis and management of this disease. According to the Standardization Uveitis Nomenclature (SUN)1 criteria, the classification of uveitis involves identifying the primary site of inflammation, time of disease onset, and characteristics of inflammation. (Tables 1 and 2; Figure 1).
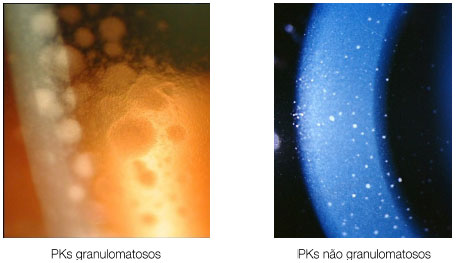
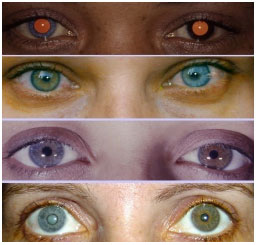
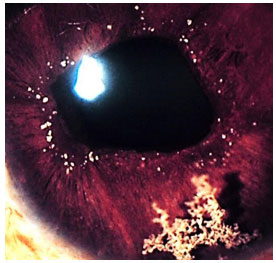
Keratic precipitates (KPs) are cellular deposits on the posterior surface of the cornea and should be carefully examined. Large (“mutton-fat”) KPs are termed granulomatous, whereas small and thin KPs are termed nongranulomatous. The presence of a grayish color and an associated fibrin meshwork between the KPs indicates recent KPs, whereas pigmentation indicates old KPs. KPs are usually located in the Arlt’s triangle, at the bottom of the cornea. However, in some uveites, KPs are diffusely distributed over the posterior surface of the cornea (Fuchs heterochromic cyclitis or sarcoidosis), whereas in herpes, they tend to be located in the affected corneal region (Figure 2)..
The activity of ocular inflammation can be measured based on the number of cells present in the anterior chamber. For this purpose, the light intensity of the slit-lamp is raised to the maximum using a 1 x 1 -mm slit, and the light beam is directed at a 45°-60° oblique angle on to the plane of the iris. (Table 3; Figures 3 and 4).



DIFFERENTIAL DIAGNOSIS
The signs and symptoms vary according to the specific cause of the uveitis and the primary site of inflammation. Patients with acute anterior uveitis often experience pain, photophobia, ocular hyperemia, and blurred vision, whereas patients with chronic anterior uveitis may only report slightly reduced visual acuity, sometimes secondary to complications such as macular edema, cataracts, or band keratopathy. Patients with intermediary uveitis mainly complain of opacities in the visual field, large floaters, and eventually decreased visual acuity. Patients with posterior uveitis frequently complain of visual loss, floaters, photopsia, metamorphopsia, scotoma, nyctalopia, or a combination of the previous complaints.
In Brazil, infection is responsible for most cases of uveitis and should always be considered first. After its exclusion, inflammation should be investigated. Diseases such as rheumatoid arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, systemic lupus erythematosus, Behçet’s disease, and sarcoidosis may have uveitis as the first and only form of ocular manifestation. Rarely, secondary causes, including trauma, masquerade syndromes with clinical manifestations such as intraocular lymphoma, or métastasés, have clinical presentations suggestive of uveitis.
FUCHS HETEROCHROMIC CYCLITIS
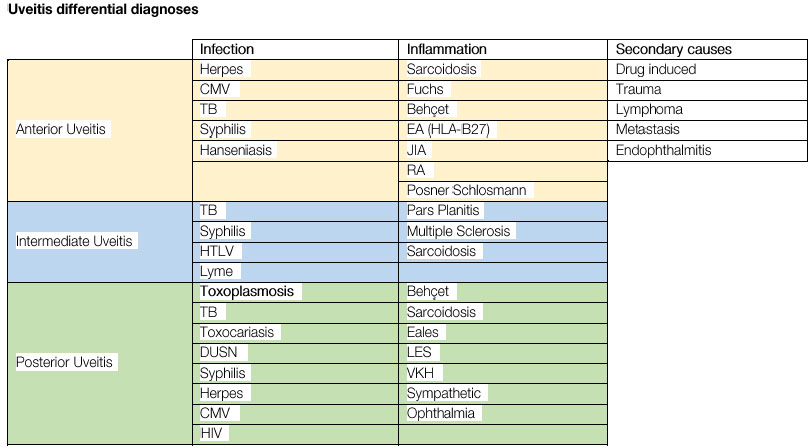
This is a nongranulomatous anterior uveitis that is chronic, insidious in onset, and unilateral in 90% of patients. It equally affects men and women. The disease manifests with the triad of uveitis + heterochromia + cataracts. The characteristic signs of the disease are as follows: the lighter-colored eye is affected; heterochromia in dark eyes is often difficult to detect, and in very light blue eyes, the darker eye is affected. The symptoms are mild, there is no significant hyperemia, and the KPs are white, diffuse, stellate, and nonconfluent. The cataracts that most frequently occur in this condition are posterior subcapsular cataracts. Filiform hemorrhage of the angle may occur after anterior chamber paracentesis (Amsler’s sign).
Glaucoma may be an associated condition in up to 20% of patients. The treatment for this condition should be individualized according to the extent of ocular inflammation, and often, treatment is not necessary. Mydriatic drugs are not necessary because synechia does not occur. Treatment with excessive topical or systemic corticoids may lead to iatrogenic events such as increased intraocular pressure and the progression of cataracts.
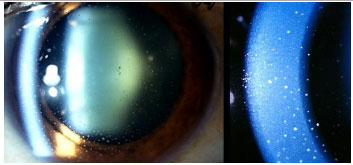
Posner-Schlossmann’s syndrome: This is a nongranulomatous, anterior, and recurrent uveitis that typically affects young male adults. It typically manifests as mild-to-moderate iritis associated with a marked increase in unilateral intraocular pressure, which can reach 60 mmHg. The angle of the anterior chamber remains open and is not associated with posterior synechiae. Ophthalmology examinations conducted between crises may show normal findings, with the exception of eventual glaucomatous optic atrophy. The treatment during a crisis includes topical steroids and antiglaucomatous drugs. Most patients have a good prognosis, but 25% of patients may progress to glaucoma.
HLA-B27-associated anterior uveitis: This is a nongranulomatous anterior uveitis, bilateral and asymmetric with synechia, and typically affects young men. It alternately affects both eyes. More than 50% of patients with HLA-B27-associated uveitis have an associated systemic condition such as ankylosing spondylitis, Reiter’s syndrome, inflammatory bowel disease, or psoriatic arthritis. In addition to ocular symptoms, patients may complain of low back pain, arthritis, psoriasis, mouth ulcers, chronic diarrhea, and urethritis. The uveitis is usually acute and recurrent but may be chronic. The symptoms in the acute phase are severe, with intense conjunctival hyperemia, photophobia, and pain. If untreated, the crisis can last for weeks up to months and is usually recurrent. An intense reaction may be observed in the anterior chamber, typically with fibrinous hypopyon, as well as posterior synechiae. The treatment includes topical or periocular corticosteroids, mydriatics-cycloplegics, and systemic corticosteroids.
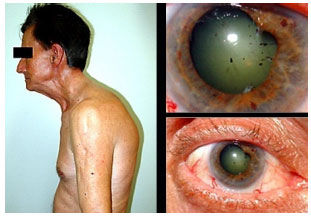
Juvenile idiopathic arthritis: This is a chronic, nongranulomatous, bilateral anterior uveitis, manifesting with synechia. It is usually asymptomatic and primarily affects girls with oligoarticular juvenile idiopathic arthritis. The patient and the family may not notice the disease because the eyes do not become externally inflamed and there is no pain. It is often only diagnosed when complications such as cataracts, band keratopathy, or glaucoma occur. The treatment includes corticosteroids, topical mydriatics, systemic corticosteroids, and immunosuppressants. Biological agents are not formally indicated for the treatment of uveitis but are often used in severe cases. The results substantially vary, and these agents have not been proven superior to steroids in the long term. Decreased vision is usually associated with amblyopia, cataracts, and band keratopathy. Intraocular lens implantation is associated with a higher frequency of complications, such as the formation of a pupillary membrane and retrolenticular opacities, and is therefore often contraindicated.
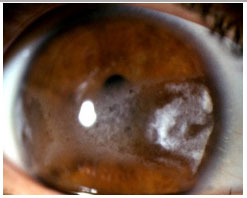
Rheumatoid arthritis: This is a bilateral, non-granulomatous anterior uveitis that typically affects middle-aged women. Ocular manifestations include scleritis, ulcerative peripheral keratitis, and anterior uveitis. The treatment consists of either topical or periocular corticosteroids, plus mydriatics. The systemic disease should always be treated according to its activity.
Traumatic uveitis: Although the severity depends on the type and intensity of the trauma, this is typically a mild-to-moderate, acute, nongranulomatous uveitis that lasts for a limited duration and responds well to the treatment with topical corticosteroids. Topical mydriatics and analgesics may be necessary. Depending on the type of trauma, the presence of an intraocular foreign body should always be suspected, particularly in unilateral chronic uveitis.
Herpes simplex: This condition involves a granulomatous or nongranulomatous anterior uveitis that is typically unilateral and recurrent and is associated with corneal edema. It manifests with KPs underlying the edema and patchy iris transillumination. It may or not be associated with keratitis and increased intraocular pressure, and it is much more symptomatic and severe in children. The development of deep corneal stromal neovascularization often occurs. The treatment includes systemic antivirals and cycloplegics-mydriatics. Herpes virus can also induce posterior uveitis, which will be described below.
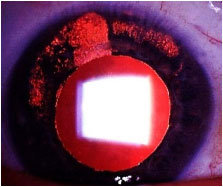
Cytomegalovirus (CMV): CMV very rarely induces unilateral, nongranulomatous anterior uveitis of a recurrent and hypertensive nature. KPs very often exhibit an amoeboid or nummular shape. This should be treated with antivirals, corticosteroids, and topical mydriatics.
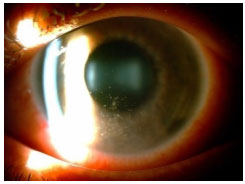
Hanseniasis: This is a chronic granulomatous disease caused by Mycobacterium leprae. The eyes are more frequently affected in Virchowian hanseniasis. Manifestations include madarosis, decreased corneal sensitivity, corneal nerve thickening, and anterior, intermediary, or posterior uveitis. Iris pearls are a characteristic sign of the disease. Systemic treatment involves rifampicin, clofazimine, and dapsone, according to the clinical stage of the disease. If uveitis appears, it should be treated with topical steroids and mydriatics.
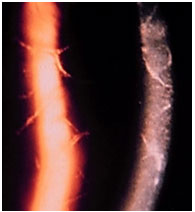
Syphilis and tuberculosis will be discussed below, since they are potential causes of all types of uveitis (anterior, intermediary, and posterior).
INTERMEDIARY UVEITIS
Intermediary uveitis classically involves the vitreous as the primary site of inflammation, but this condition often manifests with many vascular and posterior-pole changes, along with macular edema.
INTERMEDIARY UVEITIS ASSOCIATED WITH NONINFECTIOUS CAUSES
Pars planitis: Idiopathic intermediary uveitis is also termed pars planitis. This type of uveitis is most directly associated with cystoid macular edema. Inflammation is often asymptomatic and may only be detected when inflammation-related complications occur. The main symptom is floating opacities in the visual field. It is typically a chronic disease that mostly affects young women. The signs include vitreous cells, snowballs, and round vitreous condensations that are mainly located in the inferior vitreous, as well as snowbanking, a massive vitreous condensation forming a band at the inferior periphery of the retina. The treatment of mild forms consists of observation. When vision is compromised, the treatment includes periocular or intraocular steroids. Immunosuppressants are often required.
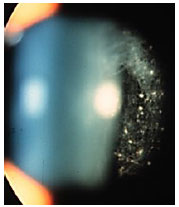
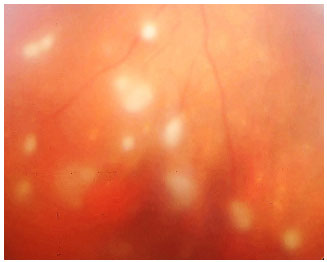

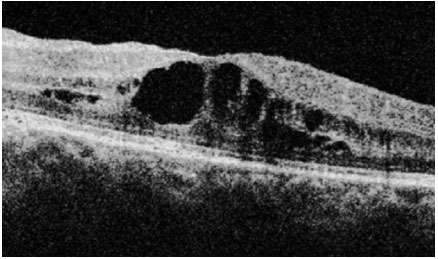
Sarcoidosis: This is a chronic systemic granulomatous disease of unknown etiology. It is less frequent in black individuals and more frequent in children and the elderly. Systemic manifestations are mainly pulmonary, and the eyes are affected in up to 50% of patients. These patients usually exhibit anergy to the purified protein derivative (PPD) test and have a previous history of erythema nodosum, skin rash, etc. The disease can affect all structures of the eye, including the eyelid and the lacrimal gland. The most common form of ocular manifestation is bilateral granulomatous uveitis (anterior, intermediary, and posterior uveitis or panuveitis). A presumed diagnosis of ocular sarcoidosis is made when there are no signs of extraocular disease and a definite diagnosis is made when non-caseating granulomas are seen in biopsy tissues. Steroids are the main treatment.
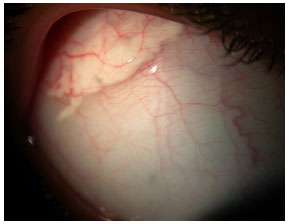
MULTIPLE SCLEROSIS
This is a demyelinating disease that mainly affects young women. It is associated with intermediary uveitis in 30% of patients, and one-fourth of these patients present with uveitis before the demyelinating disease is diagnosed. The uveitis tends to be milder than that observed in cases of pars planitis. This diagnosis should be taken into consideration because the use of biological agents, which are often prescribed to patients with intermediary uveitis that is difficult to treat, is contraindicated in patients with multiple sclerosis.
INTERMEDIARY UVEITIS ASSOCIATED WITH INFECTIOUS CAUSES
Human T-cell lymphotropic virus (HTLV): HTLV is a retrovirus that can be transmitted transplacentally, sexually, or via blood transfusion or breastfeeding. It is endemic to Japan and Brazil (mostly in the Northeast region) and is associated with spastic palsy, lymphoma, leukemia, chronic-relapsing intermediary uveitis, and retinal vasculitis. The treatment consists of steroids.
Lyme disease: There are no reports of Lyme disease in Brazil. This disease is caused by the spirochete Borrelia burgdorferi, which is endemic to the United States and some European countries, and is transmitted by a tick bite. It causes migratory erythema, arthritis, and, rarely, meningitis. The ocular manifestations include the following: follicular conjunctivitis and anterior, intermediary, or posterior uveitis, with intermediary uveitis being the most common presentation. Ocular involvement is strongly suggestive of central nervous system involvement, and the treatment consists of systemic antibiotics.
POSTERIOR UVEITIS
Posterior uveitis associated with noninfectious causes
White dot syndrome
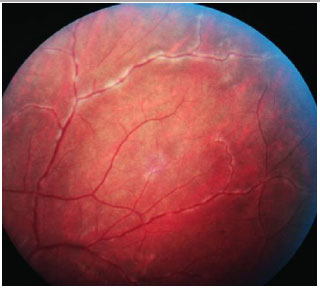
Birdshot Chorioretinopathy: This is a rare, chronic posterior uveitis with significant vitritis, without snowballs or snowbankings. It is characterized by exacerbations and remissions and mainly affects middle-aged white women. The genetic predisposition is evidenced by the association with HLA-A29. Macular edema may appear. FA hallmarks are early hypofluorescence with late hyperfluorescence. Electroretinography (ERG) is used to monitor the disease (preserved a-wave; decreased amplitude and increased latency time of b-wave). Electrooculography (EOG) may be subnormal. The treatment involves local and systemic steroids, and the use of immunosuppressive agents is often necessary.
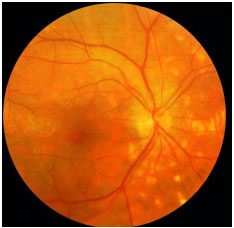
Serpiginous Choroiditis: This is a rare, chronic, bilateral posterior uveitis. It is progressive and recurrent, with months or years of inactivity between episodes. Its etiology is unknown. It typically affects middle-aged men. It is usually asymptomatic until the macula is affected. The eye is quiet, with no or minimal anterior chamber reaction, and the vitreous may exhibit fine pigmented cells. The active lesions present as grayish or yellowish subretinal infiltrates that extend outward and irregularly from the optic disc. Neovascularization of the choroid is the most common complication. This condition must be distinguished from ocular tuberculosis. Fluorescein angiography (FA): early hypofluorescence, with hyperfluorescence of the lesion margins that evolves centrally with gradual leakage in the late phase. The treatment includes local and systemic steroids. The use of immunosupressive agents is often necessary.
Multiple evanescent white dot syndrome (MEWDS): This is a transient unilateral syndrome that mainly affects young women, who present with multiple deep white dots on the outer retina or retinal pigment epithelium (RPE); granular aspect of the macula. Viral prodrome occurs in approximately 33%-50% of patients. Suddenly diminished visual acuity occurs; paracentral scotoma and relative afferent pupillary defect (RAPD) may be present. Anterior chamber reaction is minimal or absent, and vitritis is moderate. Hyperemia or edema of the optic disc may occur. It has an acute progression and spontaneously resolves in 7 weeks on an average. The loss of visual fields may persist. FA: Early hyperfluorescence and late impregnation. ERG: reduced a-wave. EOG may also be abnormal.
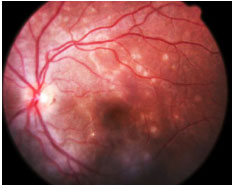
Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): This is an acute disease with multiple yellowish-white plaque-like lesions in the posterior pole, with sudden visual loss. It spontaneously resolves within a few weeks, leaving minimal scarring. Young men and women are equally affected. It is usually bilateral. One-third of patients exhibit a viral prodrome. It is associated with systemic vasculitis. FA: early hypofluorescence with late hyperfluorescence.
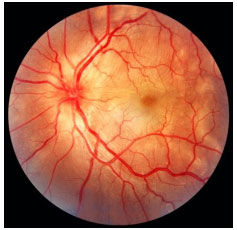
Multifocal choroiditis and panuveitis: This condition involves chronic, asymmetric, bilateral uveitis characterized by multiple chorioretinal lesions similar to those observed in presumed ocular histoplasmosis syndrome. It affects primarily elderly women. The anterior chamber reaction is mild to moderate, with nongranulomatous KPs and posterior synechia. Vitritis is moderate to severe. Fundoscopy shows yellowish-white lesions (50-100 µm in diameter) that mainly involve the choroid and outer retina. The most common cause of visual loss is choroidal neovascularization. FA: early hypofluorescence with late hyperfluorescence.
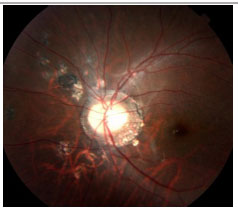
Acute Zonal Occult Outer Retinopathy (AZOOR): This condition involves an acute loss of one or more zones of the outer retina, associated with photopsia, minimal fundoscopic changes, and abnormal ERG findings. It may be unilateral or bilateral and typically affects young myopic women. Mild vitritis and a loss of visual fields may occur. FA may be normal, with only delayed retinal vascular filling. There is no consensus on treatment.
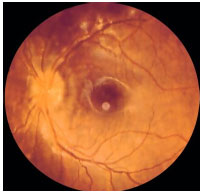
Inner Punctate Choroiditis: This is an idiopathic inflammatory disorder that affects young myopic women who complain of metamorphopsia, paracentral scotoma, photopsia, and decreased visual acuity. The lesions are small (100-200 µm) and affect the posterior pole. There is no vitritis. The lesions progress to atrophic scars and may exhibit subretinal neovascular membranes as a complication. FA: early hyperfluorescence with late hyperfluorescence.
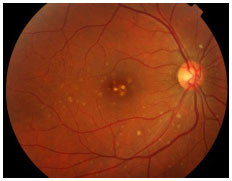
POSTERIOR UVEITIS RELATED TO INFECTIOUS CAUSES
Toxoplasmosis
This condition is caused by Toxoplasma gondii, a protozoan acquired via the ingestion of undercooked pork/chicken meat or water and food contaminated by cat feces or via the placenta. It is the main cause of posterior uveitis in Brazil and worldwide. The systemic infection may cause flu-like symptoms but is often asymptomatic.
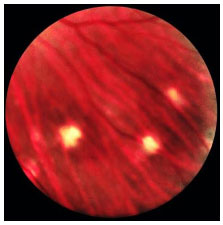
Congenital ocular toxoplasmosis occurs when the mother acquires the infection during pregnancy. The earlier it is acquired during pregnancy, the more severe the fetal infection. If the infection occurs in the first trimester, miscarriage is likely. The typical ocular lesions in the newborn are wagon-wheel macular retinochoroiditis lesions. Congenital toxoplasmosis ocular lesions can develop for years after
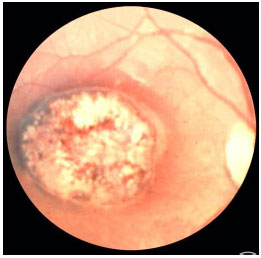
The disease is severe and can lead to permanent visual loss and legal blindness. Treatment of the infection in the newborn usually lasts for 12 months and must be prescribed in consultation with a pediatrician. After this period and depending on the severity of the disease, children should receive prophylactic treatment with 800 mg of sulfamethoxazole and 160 mg of trimethoprim for several years to avoid a subclinical crisis because children do not verbalize the symptoms. The main strategy to avoid congenital ocular toxoplasmosis is to educate mothers regarding the modes of transmission and feeding guidelines.
Acquired ocular toxoplasmosis manifests as granulomatous posterior uveitis with necrotizing exudative retinochoroiditis, typically adjacent to a scar of previous toxoplasmosis, with vitritis mainly located above the lesion (creating a “headlight in the fog” sign). Visual symptoms depend on the location of the lesion in the retina, but patients usually complain of floaters, followed by a drop in visual acuity.
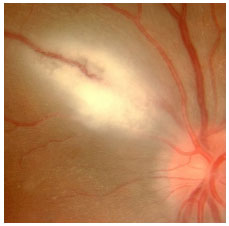
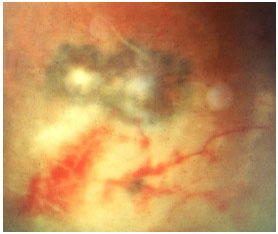
The classical treatment includes 4 g/day of sulfadiazine, 25 mg/day of pyrimethamine, and corticosteroids when necessary. It may also include a combination of 800 mg of sulfamethoxazole and 160 mg of trimethoprim. Patients who are allergic to sulfamethoxazole may receive clindamycin (300 mg orally every 6 hours) as a treatment alternative. An intravitreal injection of clindamycin and dexamethasone is also considered effective.
Patients with ocular lesions due to severe and frequently recurrent ocular toxoplasmosis may benefit from sulfamethoxazole + trimethoprim taken three times a week for several years as prophylaxis.
TUBERCULOSIS
Tuberculosis is endemic to Brazil and is a systemic granulomatous disease caused by Mycobacterium tuberculosis. It mainly presents as pulmonary tuberculosis. However, it may be disseminated via the blood and can cause infection in other organs.
Ocular manifestations are diverse and include the involvement of the eyelid, conjunctiva, iris, retina, and optic nerve. It is not clear whether the ocular manifestations are caused by the mycobacterium itself or by its immunogenic stimulus. It classically presents as a granulomatous anterior, intermediary, or posterior uveitis, the posterior form being the most common.
The PPD test is a screening test that assesses previous exposure to the mycobacterium and supports the diagnosis of ocular tuberculosis. Although it produces many false positive results, it should be interpreted as follows:
Anergic PPD (0 mm): unvaccinated patient, long period since last vaccination, or possible sarcoidosis;
PPD 0-5 mm: recently vaccinated patient;
PPD 5-10 mm: should be considered positive in immunocompromised patients, patients with human immunodeficiency virus (HIV), patients in contact with active tuberculosis, or patients with a chest X-ray consistent with tuberculosis lesions;
PPD 10-15 mm: considered positive in patients with diabetes, patients with renal failure, patients using immunosuppressants, health professionals, or immigrants from high-prevalence regions;
PPD >15 mm: considered positive in patients without risk factors.
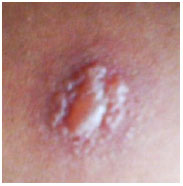
New tests based on interferons from peripheral blood lymphocytes exhibit higher specificity and sensitivity than the PPD test and lower cross-reactivity with the Bacillus Calmette-Guerin (BCG) vaccine; they also support the diagnosis of tuberculosis. However, they are costly, and their application is limited in Brazil.
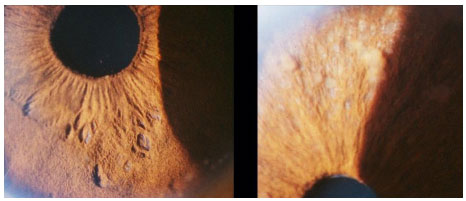
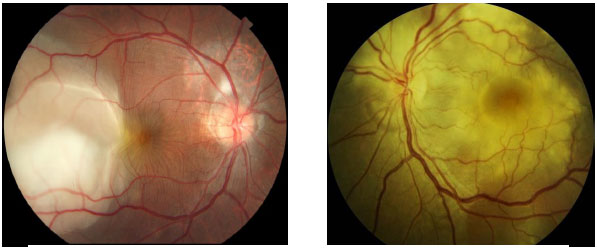
The recommended treatment for ocular tuberculosis is a quadruple therapy (rifampicin + isoniazid + pyrazinamide + ethambutol). Ethambutol should be discontinued in case of optic nerve toxicity. The Brazilian Ministry of Health recommends a treatment period of over 6 months. However, it is often prolonged up to 15 months in cases of ocular tuberculosis. The use of corticosteroids improves ocular symptoms.
SYPHILIS
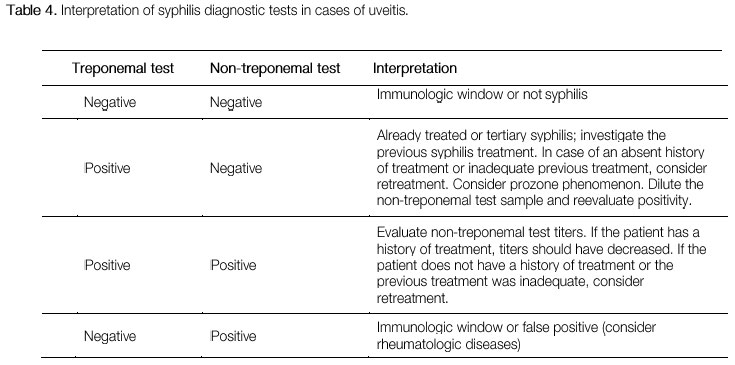
Syphilis is the great mimicker among ocular inflammatory diseases. It has a highly variable clinical spectrum and can cause all types of uveitis. Therefore, it must always be excluded in cases of ocular inflammation. The eye is affected in the secondary and tertiary stages of syphilis. The transmission mode is either sexual or via blood contact or the placenta. The diagnosis is based on treponemal tests, such as the Fluorescent Treponemal Anti body-Absorption Test (FTA-Abs) and enzyme-linked immunosorbent assay (ELISA), and non-treponemal tests, such as the Venereal Disease Research Laboratory (VDRL; Table 4). The treatment is similar to that used for neurosyphilis and consists of intravenous crystalline penicillin (4 million IU every 4 h for 14 days). The response to treatment is usually very good. However, symptoms may intensify after antibiotic administration, a condition termed the Jarisch-Herxheimer reaction. Clinical manifestations include the worsening of skin lesions, muscle pain, fever with chills, and headache. Syphilis is associated with an autoimmune reaction to the release of toxins by the bacterium after treatment. The use of systemic steroids prevents this reaction and accelerates the improvement of the signs and symptoms of uveitis.
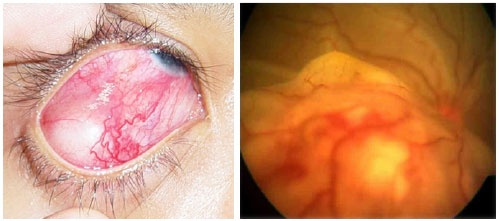
VIRAL POSTERIOR UVEITIS
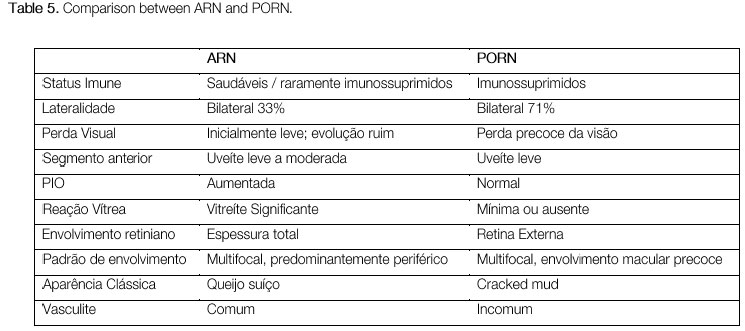
Acute retinal necrosis (ARN)
This is a rare entity characterized by acute necrosis of the entire thickness of the retina and is caused by varicella zoster virus, herpes simplex type 1 or type 2 virus, and, rarely, CMV and Epstein-Barr virus (EBV). Although ARN classically occurs in young immunocompetent individuals, recent studies have indicated that there is a predisposition in chronic users of corticosteroids and in relatively immunosuppressed patients. The clinical presentation is characterized by mild-to-moderate anterior uveitis, with increased intraocular pressure; the triad of posterior segment involvement is composed of moderate-to-severe vitritis, occlusive vasculitis, and necrotizing retinitis that initially affects the periphery of the retina and progresses rapidly and circumferentially. The second eye is affected in one-third of patients after 1-6 weeks; however, there are reports of intervals of over 30 years. The treatment should be promptly started with intravenous acyclovir (10-15 mg/kg every 8 hours for 15 days). Valacyclovir hydrochloride can also be used, with the advantage of oral administration and good bioavailability, but there are no studies comparing the drugs. With treatment, the condition usually resolves within 3 weeks. Without treatment, the course of the disease is more prolonged (approximately 12 weeks). Treatment reduces the occurrence of the disease in the contralateral eye, but the risk persists for many years. The thinning of the affected retina, associated with vitreous cell infiltration, predisposes to the development of retinal holes and ruptures, which in turn lead to rhegmatogenous retinal detachment in 75% of patients. There is no consensus with regard to early vitrectomy and the use of laser therapy in the course of ARN.
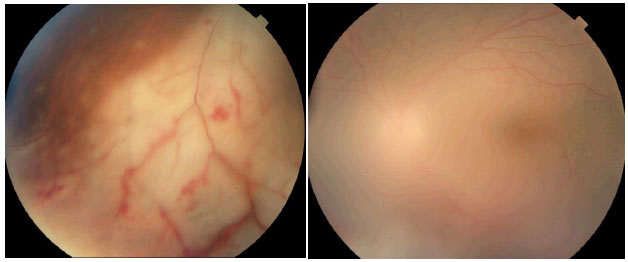
Progressive outer retinal necrosis (PORN)
PORN is a rare entity, first described in 1990. Similarly to ARN, it is caused by herpes simplex virus type 1 and 2 or by varicella zoster; however, it is mostly observed in HIV-positive individuals who are severely immunosuppressed (CD4 < 50). Typically, there is a rapidly progressing bilateral acute necrosis of the outer retina in approximately two-thirds of patients, with multifocal lesions that initially affect the posterior pole. The prognosis is very poor, and the treatment is the same as that for ARN.
CMV
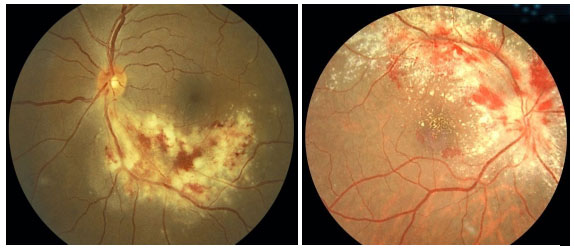
Retinitis caused by CMV is the most common eye disease associated with HIV. In the pre-highly active antiretroviral therapy (HAART) era, CMV-induced retinitis was detected in over 40% of patients. There has been a decline in its incidence, which currently ranges between 5% and 10%, depending on the country. Patients who develop CMV-induced retinitis usually have CD4 < 50/µL (mean, 17/µL) and complain of decreased visual acuity associated with floaters and a loss of visual fields; however, retinitis is asymptomatic in 15% of patients.
The clinical presentation is characterized by necrotizing retinitis without exudation and starts around the vessels and progresses centripetally, usually associated with hemorrhaging (“cheese with ketchup” lesion). Anterior involvement is mild, with little or no anterior chamber reaction, and the KPs are fine in the endothelium. Vitritis is also mild.
The clinical forms are as follows: typical or hemorrhagic, atypical or granular, and perivascular retinitis (frosted branch angiitis), the last one being the least common. As the lesions scar, the retina is left atrophic and predisposed to rhegmatogenous detachment.
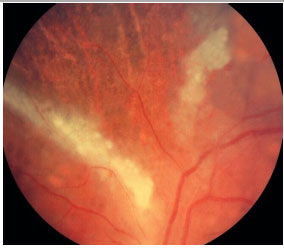
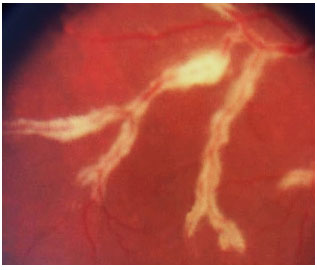
The standard treatment uses antiviral agents such as ganciclovir, foscarnet, intravenous cidofovir, and oral valganciclovir. The induction treatment lasts 2-4 weeks, and the maintenance treatment should be continued until CD4 levels rise above 150/µL The systemic administration of these drugs can cause severe side effects such as severe neutropenia secondary to ganciclovir and renal failure secondary to therapy with foscarnet and cidofovir. Intraocular ganciclovir implants are available in some countries but not in Brazil. Laser therapy can be performed in the areas predisposed to detachment. Vitrectomy via the pars plana, in association with the use of endolaser for microruptures, and the use of silicone oil are indicated when necessary. The results obtained with oil implants are better than those with gas [perfluoropropane (C3F8) or sulfur hexafluoride (SF6)].
Since the introduction of HAART, a new entity called IMMUNE RECONSTITUTION SYNDROME has appeared. This is a chronic inflammatory disorder that occurs with the improvement in the patient’s overall immunity and in CD4 levels. The clinical presentation is characterized by intense anterior uveitis and vitritis that may be associated with papillitis and cystoid macular edema. The maintenance of anti-CMV medication increases the incidence of this syndrome, especially in cases of treatment with cidofovir. The treatment consists of corticosteroids.
PAN UVEITIS
Vogt-Koyanagi-Harada (VKH) Syndrome
VKH syndrome is a rare disease of unknown etiology that involves the eye, meninges, and brain (uveo-meningo-encephalitis). Several features of the disease allow it to be described as a posterior uveitis (Harada disease) or, in most cases, as a panuveitis (VKH syndrome).
The acute presentation involves granulomatous panuveitis with bilateral serous retinal detachment, associated with headache and tinnitus. An examination of the cerebrospinal fluid may yield pleocytosis. Sometimes, retinal detachment is very extensive and may be mistaken for rhegmatogenous detachment.
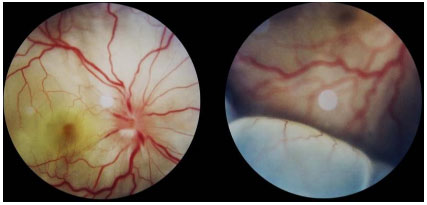
The acute disease must be aggressively treated with 1 g/day of pulse methylprednisolone for 3 days, along with prophylaxis for Strongyloides (400 mg of albendazole orally for 3 days). After pulse therapy, the patient should be maintained on oral steroids, which should be reduced according to the patient’s response. Because of its severe and relapsing nature, all patients with VKH are candidates for prolonged or chronic systemic immunosuppression. Intravitreal administration of corticosteroids rapidly reduces the uveitis and retinal detachment; however, it should always be administered in conjunction with systemic treatment.
The repeated relapses manifest as new episodes of granulomatous panuveitis associated with serous retinal detachment. In the chronic disease, fundoscopic depigmentation and RPE rearrangement, known as ’’sunset glow fundus,” are typically seen. Small yellowish spots scattered mainly in the periphery and mid-periphery of the retina, known as Dalen-Fuchs nodules, are also seen.
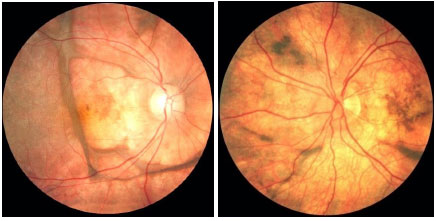
The late manifestations of the disease include skin changes such as vitiligo, poliosis, and hair loss.
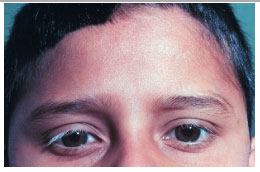
The VKH syndrome is a severe uveitis that can lead to permanent visual loss. Therefore, early diagnosis and adequate treatment are essential to minimize the damage caused by this condition.
Sympathetic ophthalmia (SO)
SO is a very rare entity that affects individuals with a history of penetrating ocular trauma, usually with the exposure of uveal tissue or previous intraocular surgery. The time between trauma and the onset of inflammation considerably varies, and this condition is often underdiagnosed. Most patients develop severe granulomatous panuveitis in the contralateral (sympathetic) eye. The symptoms range from ocular irritation and floaters to severe reduction in visual acuity. Serous retinal detachment can occur, and the disease often progresses very much like VKH syndrome, potentially leading to blindness. In addition, Dalen-Fuchs nodules are seen in the fundus in later stages of the disease. The treatment should be prompt and aggressive and should involve the use of steroids in the acute phase and subsequent systemic immunosuppression if required.
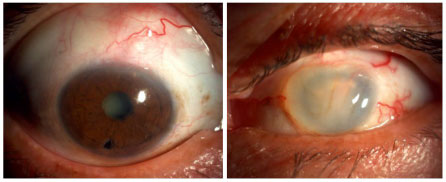
BEHCET’S DISEASE
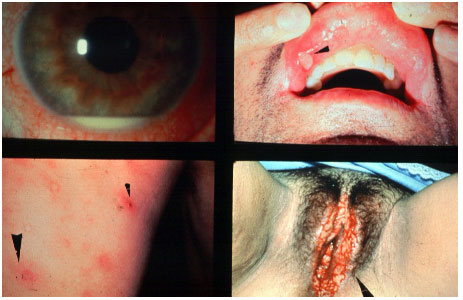
Behçet’s disease is a chronic, systemic, nongranulomatous, occlusive vasculitis of unknown cause, which is associated with HLA-B51. It is more common in Asia, the Middle East, and Brazil. This condition is clinically diagnosed based on the presence of painful mouth ulcers, genital ulcers, uveitis, and skin changes.
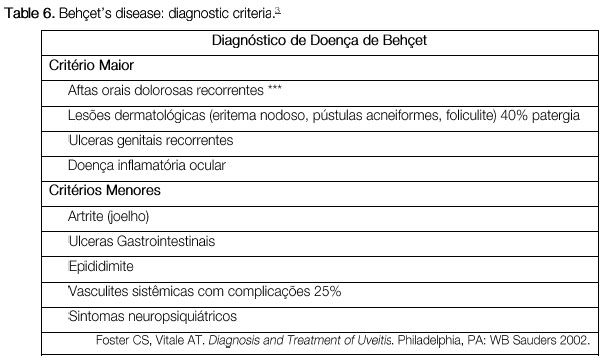
The disease is considered complete when it fulfills 4 major criteria, incomplete when it fulfills 3 major criteria or 1 major criterion plus ocular involvement, suspect when it fulfills 2 major criteria without ocular involvement, and possible when it fulfills 1 major criterion. Uveitis, which is observed in 70% of patients with systemic involvement, may be the first symptom of the disease. It is usually a bilateral panuveitis with mobile hypopyon and retinal vessel vasculitis. It may lead to permanent visual loss if not promptly and adequately treated during the crisis.
ENDOPHTHALMITIS
This condition is characterized by marked intraocular inflammation secondary to bacterial or fungal infection. In addition to the involvement of the anterior and posterior segments, keratitis or scleritis are seen. The classic signs of endophthalmitis are blurred vision, pain, conjunctival hyperemia and chemosis, eyelid edema, and hypopyon.
Endophthalmitis is classified as exogenous (acute postoperative or late traumatic) or endogenous (less common).
Endophthalmitis is clinically diagnosed. However, additional imaging exams may be necessary, especially when an intraocular foreign body is suspected. Samples of aqueous and vitreous humor should always be collected to confirm the causal agent. Treatment should be aimed at the underlying causal agent, using a topical, intravitreal, or systemic antibiotic or antifungal, along with topical corticosteroids and mydriatics. Any delay in treatment can worsen the visual prognosis and lead to blindness.
POSTOPERATIVE ACUTE ENDOPHTHALMITIS
It typically occurs in the first 6 weeks after intraocular surgery. The incidence ranges between 0.07% and 0.12%. The most reported causal agents are coagulase-negative staphylococci, but Staphylococcus aureus, Streptococcus sp, and gram-negative bacteria have also been isolated. Known preoperative risk factors include systemic immunosuppression (diabetes and cancer) and blepharoconjunctivitis. Intraoperative risk factors include capsular rupture, iris prolapse, prolonged surgery, and vitreous loss. An intense anterior chamber reaction, with significant hypopyon and vitritis, is often seen. This condition must be distinguished from toxic anterior segment syndrome (TASS), which occurs earlier (1-2 days postoperatively) and is characterized by the absence of pain and the minimal involvement of the posterior segment.
POSTOPERATIVE CHRONIC ENDOPHTHALMITIS (>6 WEEKS UP TO 2 YEARS AFTER INTRAOCULAR SURGERY)
The causal agents most commonly reported are Propionibacterium acnes, fungi, and less virulent gram-positive and gram-negative bacteria. The signs and symptoms are less severe, and the onset is more insidious than those in postoperative acute endophthalmitis. A characteristic sign is the presence of a whitish plaque in the capsular sac or the sudden appearance of hypopyon in a patient with postoperative chronic inflammation.
TRAUMATIC ENDOPHTHALMITIS
This type of endophthalmitis occurs after any penetrating ocular trauma. The most commonly associated causal agents are S. aureus and Staphylococcus epidermidis, Bacillus sp, Streptococcus sp, and several fungi. A delay of more than 12 h in surgical repair increases the risk of endophthalmitis and worsens the visual prognosis, particularly in patients with a suspected intraocular foreign body.
ENDOGENOUS ENDOPHTHALMITIS
This condition involves an intraocular inflammation secondary to blood dissemination of systemic sepsis. A complete history and a high degree of clinical suspicion are essential in these cases because infections such as endocarditis or skin and urinary tract infections can go unnoticed. The associated risk factors are systemic immunosuppression, drug use or chronic corticosteroid therapy, intravenous catheters, alcoholism, liver disease, and recent abdominal surgery. In most cases, systemic symptoms are nonspecific and include fever and myalgia. Ocular involvement is bilateral in up to 25% of patients and includes iris nodules or microabscesses, minimal or severe anterior chamber reaction (with hypopyon), whitish choroidal plaques with progressive retinal involvement, cotton-wool exudates, retinal vasculitis, and vitritis. The treatment is aimed at the underlying infection, and the identification of the causal agent is of extreme importance for the selection of the best antibiotic or antifungal therapy.
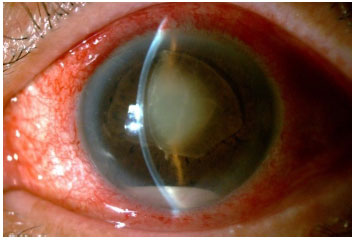
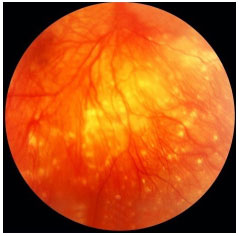
VASCULITIS
Vasculitis may be idiopathic, isolated, or associated with systemic vasculitis or an infectious or tumoral disease. The diagnosis is usually clinical, but FA is essential for the documentation and follow-up of this condition because retinal vascular involvement is usually more extensive than the fundoscopic exam suggests. The typical fundoscopic appearance is an exudation along the vessels or sheathing of the vascular walls. FA usually shows either focal or diffuse perivascular hyperfluorescence, the former in sarcoidosis and multiple sclerosis, and the latter in Behçet, Birdshot, and Eales diseases.
Arteritis is more often seen in systemic lupus erythematosus; polyarteritis nodosa; idiopathic retinal vasculitis, aneurysms, and neuroretinitis (IRVAN) syndrome; Churg-Strauss syndrome; syphilis, herpes simplex virus infection (ARN), and varicella zoster virus (PORN) infection. Phlebitis is common in sarcoidosis, multiple sclerosis, Behçet’s disease, and Eales disease. A combination of arteritis and phlebitis is usually seen in uveitis associated with toxoplasmosis, relapsing polychondritis, Wegener’s granulomatosis, Crohn’s disease, etc. When vascular inflammation is not associated with an infectious disease, the differential diagnosis can be difficult and requires a comprehensive systemic investigation. Treatment is always aimed at the underlying disease, and prolonged immunosuppression and laser photocoagulation are often necessary to avoid progression and complications such as vitreous hemorrhage, tractional retinal detachment, and neovascular glaucoma.
REFERÊNCIAS
1 A A Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveítis Nomenclature (SUN) Working Group. Standardization of uveítis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005;140:509-516. http://dx.d0i.0rg/l 0.1016/i.aio.2005.03.057
2. Oréfice e Belfort. Atlas Latino-Americano de Uveítes. Aplicativo disponível para iPhone e Ipad em AppIeStore, 2013.
3. A Foster CS, Vitale AT. Diagnosis and Treatment of Uveítis. Philadelphia, PA: WB Sauders, 2002.
LEITURA COMPLEMENTAR:
Silveira C, Belfort R Jr, Muccioli C, et al. The effect of long-term intermitent trimethoprim/sulfamethoxazole treatment on recurrences of toxoplasmic retinochoroiditis. Am J Ophthalmol. 2002:134(11:41-46 http://dx.d0i.0rg/l 0.1016/S0002-9394(02)01527-1
Nussenblatt, Robert B. Uveítis: fundamentals and clinical practice. Pennsylvania: Mosby; 2004.
Intraocular Inflammation and Uveítis 2013-2014 - Basic and Clinical Science Course - Section 9 - American Academy of Ophthalmology.
Oréfice, Fernando, Uveíte - Clínica e Cirúrgica. Cultura Médica. Segunda Edição; 2005.

Funding source: None declared.
Conflict of interest: The author declares to have no conflicts of interest.
Research Ethincs Committee Opinion: N/A.
Received on:
January 11, 2016.
Accepted on:
February 26, 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket