

Luís Expedito Sabage1,2; Vinicius Souza Pinto1; Willian Miranda Alvim1; Filipe Papa de França1; Mariana Pasqualin Wojcikiewicz3; Gabriela Bianca Manfredini2; João Pedro Vieira Neto Murta2; Felipe Domingos Ferreira Garcia2; Cleverson Teixeira Soares4,5, Josmar Sabage1,4,6
DOI: 10.17545/eOftalmo/2025.0004
ABSTRACT
This report aimed to describe a case of vitreous opacity caused by amyloid deposits in a patient with hereditary amyloidosis. A 65-year-old man with familial amyloid polyneuropathy caused by the TTR Val30Met mutation (p.Val50Met) presented with progressive visual loss in both eyes over a period of 1 year. The patient had substantial opacity and disorganization of the vitreous as well as epiretinal membrane. Ultrasound showed an attached retina with dense vitreous echoes. Pars plana vitrectomy was performed on the left eye and the vitreous material was sent for biopsy. Histological analysis of the vitreous showed the presence of amyloid deposits using Congo red staining. Familial amyloid polyneuropathy is a subtype of amyloidosis with autosomal dominant inheritance. It is associated with vitreous amyloidosis in 5.4%–35% of cases; this association is predominantly linked to the presence of a mutant variant of TTR and is observed before the age of 40 years. In the present case, the patient had a late onset of the disease but surgical treatment ensured a functional prognosis and histological analysis was essential to confirm the initial diagnostic hypothesis. This case is an example of the challenges of ophthalmic treatment in patients with rare systemic diseases. Early identification of ophthalmic changes, surgical planning, and follow-up are essential for the patient's functional recovery.
Keywords: Familial amyloid polyneuropathy; Val30Met mutation; Vitreous amyloidosis; Vitreous opacities; Vitrectomy.
RESUMO
O objetivo deste relato é descrever um caso de opacidade vítrea causada por depósitos amiloides em um paciente com amiloidose hereditária. Paciente do sexo masculino, 65 anos, com polineuropatia amiloide familiar por mutação TTR Val30Met (p.Val50Met), apresentou-se com perda visual progressiva em ambos os olhos durante um período de um ano. O paciente apresentava opacidade e desorganização substanciais do vítreo e membrana epirretiniana. O ultrassom revelou retina aplicada com material vítreo denso. Realizou-se vitrectomia via pars plana no olho esquerdo, e o material vítreo foi enviado para biópsia. A análise histológica do vítreo revelou a presença de depósitos amiloides através da coloração vermelho Congo. A polineuropatia amiloide familiar é um subtipo de amiloidose com herança autossômica dominante. A sua associação com amiloidose vítrea varia entre 5,4% e 35%, estando predominantemente ligada à presença de uma variante mutante da TTR e observada antes dos 40 anos de idade. Neste caso, o paciente apresentou um início tardio da doença, mas o tratamento cirúrgico garantiu o prognóstico funcional e a análise histológica foi essencial para confirmar a hipótese diagnóstica inicial. Este caso ilustra os desafios do tratamento oftalmológico em pacientes com doenças sistêmicas raras. A identificação precoce das alterações oftalmológicas, o planejamento cirúrgico e o acompanhamento são essenciais para a recuperação funcional do paciente.
Palavras-chave: Polineuropatia amiloide familiar; Mutação Val30Met; Amiloidose vítrea; Opacidades vítreas; Vitrectomia.
INTRODUCTION
The term amyloidosis was coined in 1854 by the German physician Rudolph Virchow after analyzing samples of amyloid bodies from brain tissue. These observations led to the assumption that the material analyzed was cellulose, which was named amyloid1. It was only years later, in 1959, with the use of electron microscopy, that Alan Cohen and Evan Calkins studied different types of amyloid deposits and, after lengthy discussions, the scientific community defined amyloid as deposits of protein material2.
Amyloidosis is a heterogeneous group of diseases caused by the extracellular deposition of insoluble fibrillar proteins, known as amyloids, which are formed from the breakdown of proteins3. Clinically, amyloidosis has two forms of manifestation: localized and systemic. In the localized form, amyloid deposits occur intracellularly and extracellularly and are restricted to the organ or tissue where the precursor protein is synthesized. In the systemic form, deposition is exclusively extracellular and occurs in organs or tissues other than the site of protein production4. The most prevalent form of systemic amyloidosis is light chain amyloidosis (AL), which is distinguished by the presence of monoclonal immunoglobulin light chains5.
Hereditary amyloidosis is another subtype of the disease, typically caused by mutant variants of proteins such as transthyretin (TTR)5. Variant hereditary transthyretin amyloidosis (ATTRv), also known as familial amyloid polyneuropathy (FAP), is an autosomal dominant systemic disease that is progressive and varies in severity. This form predominantly affects the peripheral nerves, leading to somatic and autonomic neuropathies, and can involve various organs and tissues, including the eyes6,7. Ocular involvement in ATTRv occurs in approximately 10% of patients6. TTR is a transport protein that is synthesized mainly in the liver but also in the retinal pigment epithelium (RPE). The local intraocular synthesis of mutant TTR by the RPE is clinically relevant, mainly because it explains the progression of ocular amyloidosis even after liver transplantation8.
The proteins associated with ocular amyloidosis include TTR, gelsolin, keratoepithelin, and lactoferrin, the latter two being exclusive to ocular tissue6. The main ocular manifestations of amyloidosis occur in the cornea, iridocorneal angle, and vitreous body, and are often associated with local vasculopathy and neuropathy9. The most prevalent ophthalmologic manifestations include vitreous opacities, chronic open-angle glaucoma, aberrant conjunctival vessels, keratoconjunctivitis sicca, loss of corneal sensitivity, and neurotrophic ulcers, which can result in significant visual impairment6. As mentioned above, it has been observed that the RPE synthesizes mutant TTR inside the eye, leading to the formation of amyloid deposits in the vitreous10,11. In this article, we describe a case of late onset vitreous amyloidosis in a patient with ATTRv and review the main aspects related to this ocular disease.
CASE REPORT
A 65-year-old patient with FAP and TTR missense variant p.Val30Met (c.148G>A; RefSeq: NM_000371.4), which results in the exchange of the p.Val50Met protein, presented with progressive visual loss in both eyes over a period of 1 year. Upon examination, the patient had best corrected visual acuity (BCVA) of 20/80 in the right eye (OD) and 20/400 in the left eye (OS), with IOP of 12 mmHg in both eyes, clear conjunctiva, transparent cornea, anterior chamber without reaction, and mild nuclear cataract. Retinal mapping showed opacity and disorganization of the vitreous (OD +2/4, OS +4/4). Complementary tests were also performed, and spectral-domain optical coherence tomography (SD-OCT) showed an epiretinal membrane (ERM) in the OD (Figure 1). The OS, however, could not be assessed by SD-OCT due to media opacity. Similarly, ultrasound of the eye showed an attached retina with dense vitreous material and posterior vitreous detachment.
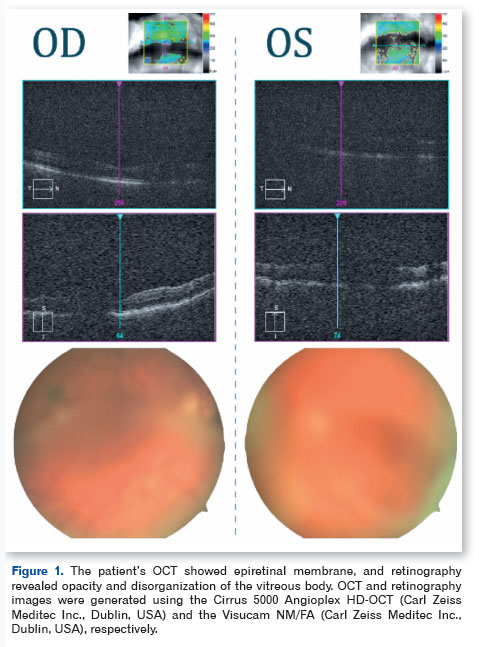
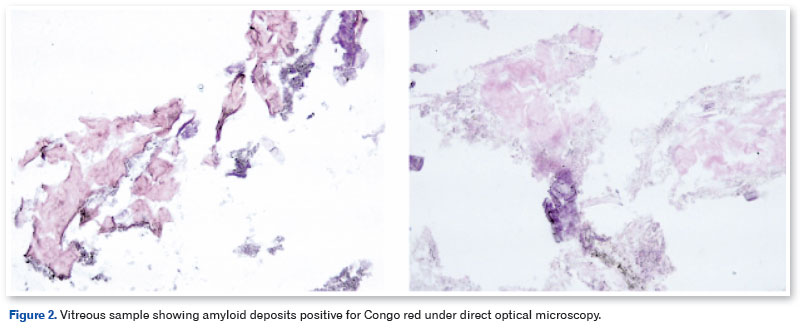
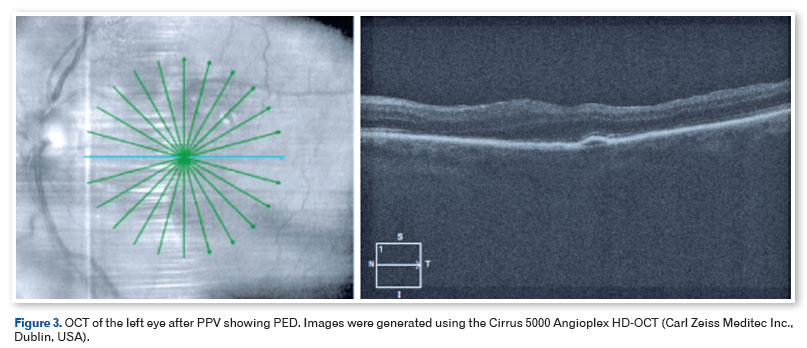
The patient underwent pars plana vitrectomy (PPV) with C3F8 13%, removal of the ERM, and peeling of the internal limiting membrane (ILM) with membrane blue dye in the OS, and the vitreous material was sent for biopsy. There were no intraoperative complications. Histological analysis of the vitreous body showed the presence of amyloid deposits through Congo red staining observed under direct light (Figure 2). Three months after surgery, the patient presented with 20/40 OE BCVA, and another SD-OCT showed preservation of the anatomy of the retinal layers and detachment of the RPE (PED) without clinical significance (Figure 3). The patient was then referred for phacoemulsification with an OS intraocular lens implant, with a final BCVA of 20/20. RPE detachment is being monitored periodically and no change/increase was observed after 6 months.
DISCUSSION
Familial amyloid polyneuropathy and vitreous amyloidosis
As previously described, vitreous amyloidosis is one of the most characteristic ocular manifestations of ATTRv and, in most cases, presents mutations of the TTR gene in p.Val50Met (Val30Met), p.Arg54Gly (Arg34Gly), p.Tyr134Cys (Tyr114Cys), p.Thr69Ala (Thr49Ala), and p.Glu74Lys (Glu54Lys)6,12-16. Vitreous amyloidosis is typically bilateral and asymmetrical, and is observed predominantly before the age of 40 years6. ATTRv due to the TTR Val30Met mutation can present with late onset, occurs usually after the age of 50 years, and accounts for approximately 35% of cases17. In these patients, diagnosis is often late, with an average interval of 3.8 years from the onset of symptoms, due to the lack of family history, phenotypic variability, and the similarity to other causes of neuropathy, such as diabetes or vitamin deficiency17,18. The presence of monoclonal gammopathy can further induce error in clinical suspicion of AL amyloidosis, making diagnosis more difficult19.
From a neurological perspective, late presentation is associated with greater severity of the disease at the time of diagnosis, including greater neurological impairment and pronounced autonomic involvement17,20. Vitreous opacities are described as the most common ocular alteration in patients with late onset disease and occur in more than half of the eyes examined in some studies21. In isolated cases, they may even be the first detectable clinical manifestation22. However, recent studies indicate that even among patients with established neurological symptoms, vitreous opacities may be absent on initial assessment, which suggests that ocular involvement may be underestimated and appear at a later stage22.
Clinical characteristics
It has been observed that these opacities form in the peripheral vitreous and extend centripetally23. The collagen fibers of the vitreous act as the basis for the aggregation of mutant TTR. This protein has a high degree of affinity with the basal membranes and the vitreous matrix. It is mainly composed of type II collagen and is structurally and biochemically similar to the collagen of the basal membranes, which favors amyloid deposition. Thus, vitreous opacities are characterized by a classic appearance, described as resembling leaves, films, bands, spider webs, cotton, fine fibers, or glass wool, the last being most often related to the disease6.
Vitreous opacities are caused by cell debris, proteins, blood, deposits, or inflammatory exudates suspended in the vitreous humor; these causes present different patterns that are important for their clinical identification. We highlight the following opacities: vitreous syneresis, which has transparent, mobile, and floating filaments with eye movement; uveitis, which has denser opacities that reduce visual acuity; and vitreous hemorrhage, which has a characteristic dark red color24. Asteroid hyalosis is another vitreous opacity that is commonly found; it is mostly benign and asymptomatic and is characterized by bright yellowish-white dots adhered to the vitreous matrix25,26. Eye ultrasound is an important diagnostic tool in these cases27.
In ATTRv, chorioretinopathy has been described as a potential ocular manifestation, often involving vascular changes such as choroidal vascular attenuation or choroidal vascular occlusion28. However, in this patient, despite the advanced disease and the prominent deposition of amyloid in the vitreous, no vascular alterations were observed on fundoscopic or angiographic examination. This absence of vascular involvement of the choroid suggests that ocular amyloid deposition can progress to a severe structural deficiency without necessarily inducing obvious vascular disease; it also suggests that such changes may be subclinical, thus making current diagnostic detection difficult29. Furthermore, long-term follow-up suggests that visual function may be intact in the first few years after detection and that visual deterioration may occur over several years29. Therefore, multimodal evaluation is recommended even in the absence of initial fundoscopic changes.
Surgical treatment
The standard treatment for this condition is PPV with peeling of the ILM but opacities can relapse due to the dispersion of residual deposits or the continuous intraocular production of mutant TTR30-32. Although PPV significantly improves visual acuity, the recurrence of opacities, the presence of worsening sensorimotor neuropathy, severe autonomic dysfunction, and intraoperative complications are challenges for the long-term treatment of these patients33-35. In this case, the absence of recurrence after 6 months suggests the complete removal of all vitreous parts during PPV. However, late recurrence is still possible due to synthesis of mutant TTR in the RPE6.
After surgery, this patient's cataract evolved rapidly, which is also to be expected after PPV given that cataract formation is the most common complication of PPV. The incidence of cataract surgery after PPV is estimated at approximately 40% and can increase to up to 85% in 5 years36,37. Considering this, the combined technique of PPV with phacoemulsification in a single procedure has been increasingly used with safety38.
Future treatments
Gene therapy is a promising future treatment strategy for TTR amyloidosis, focusing mainly on strategies to suppress the expression of the variant TTR gene by degrading the TTR messenger RNA39,40. The mechanism of action of tafamidis, one of the anti-amyloid drugs, is to stabilize mutant TTR by binding to T4 binding sites of the TTR tetramer, thereby preventing the dissociation of the latter into amyloidogenic and toxic monomers and intermediates39. Indications for use depend on the stage of the disease, age of the patient, associated illnesses, severity of heart disease, and contraindications. Tafamidis is preferred for the treatment of early-stage symptomatic FAP. The neuropathy must be symptomatic, with an amyloidogenic TTR mutation, biopsy-proven amyloidosis, and stage I disease. In a double-blind randomized study, there was no progression of neuropathy in 60% of patients in the tafamidis group versus 38% in the placebo group39.
Another treatment option is patisiran (ALN-TTR02), which consists of a specific siRNA for TTR mRNA formulated in LNPs and that shows a robust, dose-dependent and statistically significant reduction in serum TTR protein levels40,41. According to the APOLLO study42, patisiran improves neurological symptoms in patients receiving dual therapy, without having relevant adverse effects. There is another medication available, inotersen, which was recently approved by the US FDA for the treatment of polyneuropathy associated with amyloidosis. By targeting TTR mRNA transcripts, inotersen decreases translation and protein synthesis, thus reducing the deposition of misfolded proteins. Inotersen and patisiran have shown excellent results in hATTR patients with polyneuropathy. Inotersen and patisiran slowed the progression of the disease and even potentially reversed it43.
Histological analysis
Congo red dye under polarized light is the main histological method used to identify amyloid deposits44. However, because of the high concentration of deposits in this patient, direct light was sufficient to identify the material. Although it does not accurately determine the amyloid nature of protein aggregates, it remains the fastest and most practical method for staining amyloids in vitro44,45. In this case, the patient had a late onset of the disease, which required the exclusion of other etiologies for the vitreous opacities. In addition to surgical treatment guaranteeing a functional prognosis, histology was essential to confirm the diagnostic hypothesis and exclude other causes of vitreous opacity.
Other stains can also be used to detect these deposits. Thioflavin T is a fluorescent dye that binds to amyloid fibrils; after binding, its fluorescence increases significantly, emitting a greenish light that can be visualized under a fluorescence microscope. Another dye that can be used identify them is hematoxylin and eosin (H&E). Although H&E is not specific for amyloids, it is the most widely used stain in general histopathology, and amyloid proteins usually appear as homogeneous pink deposits on the slide. This technique is not conclusive on its own, and therefore, is not recommended as the main method for identifying amyloids44,46.
This case is an example of the ophthalmic treatment challenges in patients with rare systemic diseases. The vitreous is an important site of amyloid deposition, which often leads to progressive visual impairment requiring early surgical treatment. PPV, when properly indicated, remains the main treatment and provides diagnostic confirmation and functional recovery. Ultimately, multidisciplinary follow-up is essential because early ophthalmologic assessment and timely intervention can preserve visual function and improve the overall quality of life of individuals with systemic amyloidosis. This case highlights the importance of early ophthalmologic evaluation in patients with hATTR. Vitreous amyloidosis can be the initial or predominant ocular finding, and PPV allows diagnostic confirmation and visual recovery. Thus, comprehensive, multidisciplinary monitoring is essential in the treatment of ocular and systemic manifestations of ATTRv.
ACKNOWLEDGMENTS
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo through author LES (number 2024/16042-9).
REFERENCES
1. Sipe JD, Cohen AS. Review: History of the Amyloid Fibril. J Struct Biol. 2000;130(2-3):88-98.
2. Cohen AS, Calkins E. Electron Microscopic Observations on a Fibrous Component in Amyloid of Diverse Origins. Nature. 1959;183(4669):1202-3.
3. Vaxman I, Gertz M. When to Suspect a Diagnosis of Amyloidosis. Acta Haematol. 2020;143(4):304-311.
4. Blancas-Mejía LM, Ramirez-Alvarado M. Systemic Amyloidoses. Annu Rev Biochem. 2013 Jun 2;82(1):745-74.
5. Comenzo RL. Amyloidosis. Curr Treat Options Oncol. 2006;7(3):225-36.
6. Minnella AM, Rissotto R, Antoniazzi E, Di Girolamo M, Luigetti M, Maceroni M, et al. Ocular Involvement in Hereditary Amyloidosis. Genes (Basel). 2021;12(7):955.
7. Carroll A, Dyck PJ, de Carvalho M, Kennerson M, Reilly MM, Kiernan MC, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2022;93(6):668-678.
8. Munar-Qués M, Salvá-Ladaria L, Mulet-perera P, Solé M, López-Andreu FR, Saraiva MJM. Vitreous amyloidosis after liver transplantation in patients with familial amyloid polyneuropathy: Ocular synthesis of mutant transthyretin. Amyloid. 2000;7(4):266-9.
9. Kiuru-Enari S, Haltia M. Hereditary gelsolin amyloidosis. Handb Clin Neurol. 2013;115:659-81.
10. Kawaji T, Ando Y, Ando E, Nakamura M, Hirata A, Tanihara H. A case of vitreous amyloidosis without systemic symptoms in familial amyloidotic polyneuropathy. Amyloid. 2004;11(4):257-9.
11. Salvador F, Mateo C, Alegre J, Reventos A, García-Arumi J, Corcostegui B. Vitreous amyloidosis without systemic or familial involvement. Int Ophthalmol. 1993;17(6):355-7.
12. Togashi S, Watanabe H, Nagasaka T, Shindo K, Shiozawa Z, Maeda S, et al. An aggressive familial amyloidotic polyneuropathy caused by a new variant transthyretin Lys 54. Neurology. 1999;53(3):637-9.
13. Mazzeo A, Russo M, Di Bella G, Minutoli F, Stancanelli C, Gentile L, et al. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J Neuromuscul Dis. 2015;2(s2):S39-S48.
14. Ueno S, Uemichi T, Yorifuji S, Tarui S. A novel variant of transthyretin (Tyr114 to Cys) deduced from the nucleotide sequences of gene fragments from familial amyloidotic polyneuropathy in Japanese sibling cases. Biochem Biophys Res Commun. 1990;169(1):143-7.
15. Levy J, Hawkins PN, Rowczenio D, Godfrey T, Stawell R, Zamir E. Familial amyloid polyneuropathy associated with the novel transthyretin variant Arg34Gly. Amyloid. 2012;19(4):201-3.
16. Martin SE, Benson MD, Hattab EM. The pathologic spectrum of oculoleptomeningeal amyloidosis with Val30Gly transthyretin gene mutation in a postmortem case. Hum Pathol. 2014;45(5):1105-8.
17. Waddington-Cruz M, Wixner J, Amass L, Kiszko J, Chapman D, Ando Y; THAOS investigators. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. 2021;10(2):753-66.
18. Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63(1):129-38.
19. Correia AS, Mendonça M, Caetano A, Medeiros E. A sporadic case of late-onset familial amyloid polyneuropathy with a monoclonal gammopathy. Neuromuscul Disord. 2015;25(8):658-60.
20. Obi CA, Mostertz WC, Griffin JM, Judge DP. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist Debakey Cardiovasc J. 2022;18(2):17-26.
21. Ferreira N, Dias D, Coelho T. Specific ophtalmologic changes in late onset familial amyloid polyneuropathy (FAP) portuguese patients. Orphanet J Rare Dis. 2015 Nov 2;10(Suppl 1):P63.
22. Gondim FAA, Holanda Filha JG, Moraes Filho MO. Ophthalmological manifestations of hereditary transthyretin amyloidosis. Arq Bras Oftalmol. 2022;85(5):528-538.
23. Mathew R, Zeppieri M. Ocular Amyloidosis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025 Jan. 2024 May 28.
24. Milston R, Madigan MC, Sebag J. Vitreous floaters: Etiology, diagnostics, and management. Surv Ophthalmol. 2016;61(2):211-27.
25. Zaher E, Blumenthal Y, Blumenthal EZ. The lack of floater perception in eyes with asteroid hyalosis and its direct implications on laser vitreolysis. Int J Retina Vitreous. 2024;10(1):80.
26. Snehi S, Bang PH, Mamtani N, Singh K. Galactic vision: asteroid hyalosis in vitreous with moonlit optic disc. BMJ Case Rep. 2024;17(5):e260387.
27. Budhram G, Cronsell J, Schroeder M, Sautner J, Schoenfeld E, Elia T, et al. Mobile vitreous opacities on ocular ultrasonography are not always pathologic: a cross-sectional survey in an asymptomatic population. Am J Emerg Med. 2015;33(12):1808-13.
28. Roybal CN, Sanfilippo CJ, Nazari H, Law JC, Bhaleeya S, Chui Ming GC, et al. multimodal imaging of the retina and choroid in systemic amyloidosis. Retin Cases Brief Rep. 2015;9(4):339-46.
29. Nagura K, Inoue T, Ching J, Sato A, Kitahata S, Maruyama-Inoue M, et al. Long-term follow-up of a case of amyloidosis-associated chorioretinopathy. Am J Ophthalmol Case Rep. 2020 Sep 2;19:100846.
30. Venkatesh P, Selvan H, Singh SB, Gupta D, Kashyap S, Temkar S, et al. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017;124(7):1014-1022.
31. Ferreira NN, Dias DAC, Carvalho RPA, Coelho MTPM. Re-intervention in de novo vitreous opacities after pars plana vitrectomy in familial amyloidotic polyneuropathy TTR VAL30METPORTUGUESE PATIENTS. Retin Cases Brief Rep. 13(3):273-8.
32. Beirão NM, Matos E, Beirão I, Costa PP, Torres P. Recurrence of vitreous amyloidosis and need of surgical reintervention in Portuguese patients with familial amyloidosis ATTR V30M. Retina. 2011;31(7):1373-7.
33. Xu Q, Wang X, Zhang Z, Li J, Liu H, Ji S, et al. Case Report: Vitreous Amyloidosis Caused by a TTR Lys55Asn Mutation With Intraoperative Suprachoroidal Hemorrhage. Front Med (Lausanne). 2022 Jan 24;8:797223.
34. Xue YW, Xiao YQ. Vitreous amyloidosis caused by Lys55Asn mutation in TTR with peripheral neuropathy onset: a case report of FAP-related complications. Int J Ophthalmol. 2025;18(1):184-186.
35. Tan Y, Tao Y, Sheng YJ, Zhang CM. Vitreous amyloidosis caused by a Lys55Asn variant in transthyretin: A case report. World J Clin Cases. 2022;10(32):12000-12006.
36. Jackson TL, Donachie PHJ, Sparrow JM, Johnston RL. United Kingdom National Ophthalmology Database Study of Vitreoretinal Surgery: report 1; case mix, complications, and cataract. Eye (Lond). 2013;27(5):644-51.
37. Soliman MK, Hardin JS, Jawed F, Uwaydat SH, Faramawi MF, Chu CJ, et al. A Database Study of Visual Outcomes and Intraoperative Complications of Postvitrectomy Cataract Surgery. Ophthalmology. 2018;125(11):1683-91.
38. Farahvash A, Popovic MM, Eshtiaghi A, Kertes PJ, Muni RH. Combined versus Sequential Phacoemulsification and Pars Plana Vitrectomy: A Meta-Analysis. Ophthalmol Retina. 2021;5(11):1125-1138.
39. Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6(2):129-39.
40. Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086-97.
41. Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015 Dec 4;10:109.
42. Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21.
43. Robinson C, Pham C, Zamarripa AM, Dugay CS, Lee CA, Berger AA, et al. Inotersen to Treat Polyneuropathy Associated with Hereditary Transthyretin (hATTR) Amyloidosis. Health Psychol Res. 2022;10(5):67910.
44. Yakupova EI, Bobyleva LG, Vikhlyantsev IM, Bobylev AG. Congo Red and amyloids: history and relationship. Biosci Rep. 2019;39(1):BSR20181415.
45. Howie AJ, Brewer DB, Howell D, Jones AP. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest. 2008;88(3):232-42.
46. Xue C, Lin TY, Chang D, Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R Soc Open Sci. 2017;4(1):160696.
Funding: No specific financial support was available for this study.
Approved by the following research ethics committee: Universidade Positivo (número 5.504.706).
Conflict of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
November 3, 2025.
Accepted on:
January 16, 2026.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados









 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket