

Luís Expedito Sabage1,2; Vinicius Souza Pinto1; Willian Miranda Alvim1; Filipe Papa de França1; Mariana Pasqualin Wojcikiewicz3; Gabriela Bianca Manfredini2; João Pedro Vieira Neto Murta2; Felipe Domingos Ferreira Garcia2; Cleverson Teixeira Soares4,5, Josmar Sabage1,4,6
DOI: 10.17545/eOftalmo/2025.0004
RESUMO
O objetivo deste relato é descrever um caso de opacidade vítrea causada por depósitos amiloides em um paciente com amiloidose hereditária. Paciente do sexo masculino, 65 anos, com polineuropatia amiloide familiar por mutação TTR Val30Met (p.Val50Met), apresentou-se com perda visual progressiva em ambos os olhos durante um período de um ano. O paciente apresentava opacidade e desorganização substanciais do vítreo e membrana epirretiniana. O ultrassom revelou retina aplicada com material vítreo denso. Realizou-se vitrectomia via pars plana no olho esquerdo, e o material vítreo foi enviado para biópsia. A análise histológica do vítreo revelou a presença de depósitos amiloides através da coloração vermelho Congo. A polineuropatia amiloide familiar é um subtipo de amiloidose com herança autossômica dominante. A sua associação com amiloidose vítrea varia entre 5,4% e 35%, estando predominantemente ligada à presença de uma variante mutante da TTR e observada antes dos 40 anos de idade. Neste caso, o paciente apresentou um início tardio da doença, mas o tratamento cirúrgico garantiu o prognóstico funcional e a análise histológica foi essencial para confirmar a hipótese diagnóstica inicial. Este caso ilustra os desafios do tratamento oftalmológico em pacientes com doenças sistêmicas raras. A identificação precoce das alterações oftalmológicas, o planejamento cirúrgico e o acompanhamento são essenciais para a recuperação funcional do paciente.
Palavras-chave: Polineuropatia amiloide familiar; Mutação Val30Met; Amiloidose vítrea; Opacidades vítreas; Vitrectomia.
ABSTRACT
This report aimed to describe a case of vitreous opacity caused by amyloid deposits in a patient with hereditary amyloidosis. A 65-year-old man with familial amyloid polyneuropathy caused by the TTR Val30Met mutation (p.Val50Met) presented with progressive visual loss in both eyes over a period of 1 year. The patient had substantial opacity and disorganization of the vitreous as well as epiretinal membrane. Ultrasound showed an attached retina with dense vitreous echoes. Pars plana vitrectomy was performed on the left eye and the vitreous material was sent for biopsy. Histological analysis of the vitreous showed the presence of amyloid deposits using Congo red staining. Familial amyloid polyneuropathy is a subtype of amyloidosis with autosomal dominant inheritance. It is associated with vitreous amyloidosis in 5.4%–35% of cases; this association is predominantly linked to the presence of a mutant variant of TTR and is observed before the age of 40 years. In the present case, the patient had a late onset of the disease but surgical treatment ensured a functional prognosis and histological analysis was essential to confirm the initial diagnostic hypothesis. This case is an example of the challenges of ophthalmic treatment in patients with rare systemic diseases. Early identification of ophthalmic changes, surgical planning, and follow-up are essential for the patient's functional recovery.
Keywords: Familial amyloid polyneuropathy; Val30Met mutation; Vitreous amyloidosis; Vitreous opacities; Vitrectomy.
INTRODUÇÃO
O termo amiloidose surgiu em 1854 pelo médico alemão Rudolph Virchow após análise de amostras de corpos amiláceos cerebrais. Essas observações levaram a suposição de que o material analisado era celulose, nomeando-o amiloide1. Somente anos mais tarde, em 1959, com o uso da microscopia eletrônica, Alan Cohen e Evan Calkins estudaram diferentes tipos de depósitos amiloides e, após longas discussões, a comunidade científica definiu o amiloide como depósitos de material proteico2.
A amiloidose é um grupo heterogêneo de doenças causadas pela deposição extracelular de proteínas fibrilares insolúveis, conhecidas como amiloides, que se formam a partir do desdobramento de proteínas3. Clinicamente, a amiloidose manifesta-se de duas formas distintas: localizada e sistêmica. Na forma localizada, os depósitos de amiloide ocorrem tanto intracelularmente como extracelularmente, restritos ao órgão ou tecido onde a proteína precursora é sintetizada. Na forma sistêmica, a deposição é exclusivamente extracelular e ocorre em órgãos ou tecidos diferentes do local de produção da proteína4. A forma mais prevalente de amiloidose sistêmica é a amiloidose de cadeia leve (AL), que se distingue pela presença de cadeias leves de imunoglobulina monoclonal5.
A amiloidose hereditária abrange um outro subtipo da doença, tipicamente causada por variantes mutantes de proteínas, como a transtirretina (TTR)5. A variante amiloidose hereditária por transtirretina (vATTR), também conhecida como polineuropatia amiloide familiar (FAP), é uma doença sistêmica autossômica dominante que é progressiva e varia em gravidade. Esta forma afeta predominantemente os nervos periféricos, levando a neuropatias somáticas e autonômicas, e pode envolver vários órgãos e tecidos, incluindo os olhos6,7. O envolvimento ocular na vATTR ocorre em aproximadamente 10% dos pacientes6. A TTR é uma proteína de transporte sintetizada principalmente no fígado, mas pode também ser sintetizada no epitélio pigmentar da retina (EPR). A síntese intraocular local de TTR mutante pelo EPR é clinicamente relevante, principalmente porque explica a progressão da amiloidose ocular mesmo após o transplante hepático8.
Entre as proteínas associadas à amiloidose ocular, destacam-se a TTR, gelsolina, queratoepitelina e lactoferrina, sendo essas duas últimas exclusivas do tecido ocular6. As principais manifestações oculares da amiloidose ocorrem na córnea, no ângulo iridocorneano e no vítreo, frequentemente associadas a vasculopatias e neuropatias locais9. As manifestações oftalmológicas mais prevalentes abrangem opacidades vítreas, glaucoma crônico de ângulo aberto, vasos conjuntivais aberrantes, ceratoconjuntivite seca, perda de sensibilidade da córnea e úlceras neurotróficas, que podem resultar em deficiência visual significativa6. Conforme mencionado, em certos casos, observa-se que o EPR sintetiza TTR mutante dentro do olho, levando à formação de depósitos amiloides no vítreo10,11. Neste artigo, descrevemos um caso de amiloidose vítrea de início tardio em um paciente com vATTR e revisamos os principais aspectos relacionados à doença ocular.
RELATO DE CASO
Paciente de 65 anos com FAP e variante missense TTR p.Val30Met (c.148G>A; RefSeq: NM_000371.4), o que resulta na troca da proteína p.Val50Met, apresentou-se com perda visual progressiva em ambos os olhos durante um período de um ano. Ao exame, o paciente apresentava acuidade visual corrigida (AVCC) no olho direito (OD) de 20/80 e no olho esquerdo (OE) de 20/400, com pressão intraocular de 12 mmHg em ambos os olhos, conjuntiva clara, córnea transparente, câmara anterior sem reação e catarata nuclear leve. O mapeamento da retina revelou opacidade e desorganização do vítreo (OD +2/4, OE +4/4). Ademais, prosseguiu-se para realização de exames complementares, a tomografia de coerência óptica spectral domain (OCT-SD) revelou membrana epirretiniana (MER) no OD (Figura 1). Já o OE não pôde ser avaliado na OCT-SD devido à opacidade de meios. De modo semelhante, o ultrassom ocular revelou retina aplicada com material vítreo denso e descolamento do vítreo posterior (DVP).
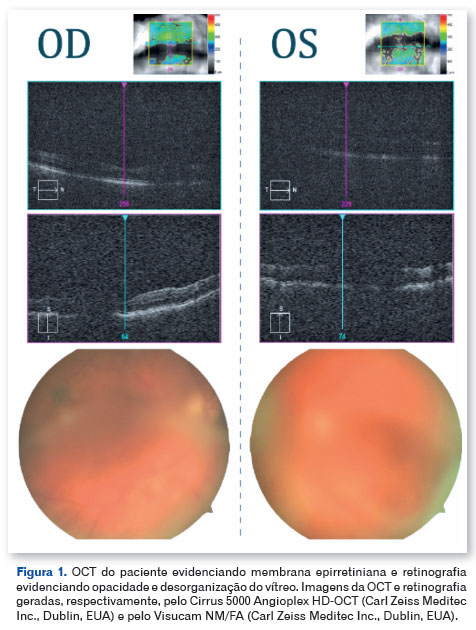
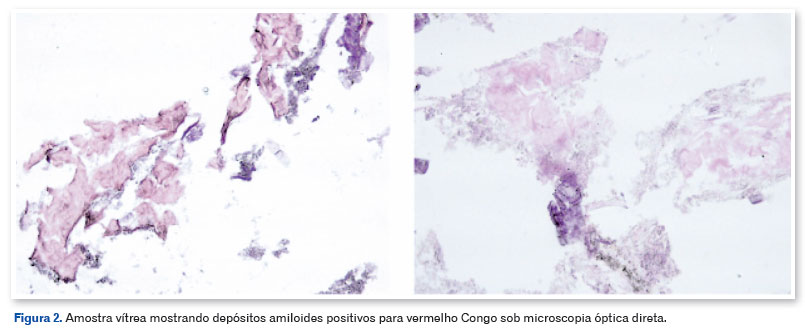
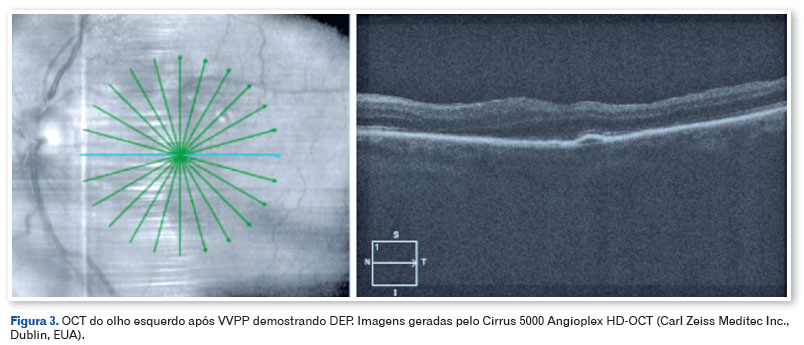
O paciente foi submetido à vitrectomia via pars plana (VVPP) com C3F8 13%, remoção da MER e peeling da membrana limitante interna (MLI) com corante membrane blue no olho esquerdo e o material vítreo foi enviado para biópsia. Não houve complicações intraoperatórias. A análise histológica do vítreo revelou a presença de depósitos amiloides através da coloração vermelho Congo observada sob luz direta (Figura 2). Três meses após a cirurgia, o paciente apresentava-se com AVCC OE 20/40, a nova OCT-SD mostrou preservação da anatomia das camadas da retina e descolamento do EPR (DEP) sem significância clínica (Figura 3). O paciente foi então encaminhado para facoemulsificação com implante de lente intraocular em OE, apresentando uma AVCC final de 20/20. O descolamento do EPR está sendo monitorizado periodicamente e não apresentou alteração/aumento após seis meses.
DISCUSSÃO
Polineuropatia amiloide familiar e amiloidose vítrea
Como descrito anteriormente, a amiloidose vítrea é uma das manifestações oculares mais características da vATTR e, na maioria dos casos, apresenta mutações do gene TTR em p.Val50Met (Val30Met), p.Arg54Gly (Arg34Gly), p.Tyr134Cys (Tyr114Cys), p.Thr69Ala (Thr49Ala) e p.Glu74Lys (Glu54Lys)6,12-16. A amiloidose vítrea é tipicamente bilateral, assimétrica e observada predominantemente antes dos 40 anos de idade6. A vATTR devido à mutação TTR Val30Met pode apresentar-se com início tardio, normalmente após os 50 anos de idade, e é responsável por aproximadamente 35% dos casos17. Nestes pacientes, o diagnóstico é frequentemente tardio, com um intervalo médio de 3,8 anos desde o início dos sintomas, devido à ausência de antecedentes familiares, variabilidade fenotípica e semelhança com outras causas de neuropatia, tais como diabetes ou deficiência vitamínica17,18. A presença de gamopatia monoclonal pode induzir ainda mais o erro na suspeita clínica em relação à amiloidose AL, dificultando o diagnóstico19.
Do ponto de vista neurológico, a apresentação tardia está associada a uma maior gravidade da doença no momento do diagnóstico, incluindo maior comprometimento neurológico e envolvimento autonômico pronunciado17,20. As opacidades vítreas são descritas como a alteração ocular mais comum em pacientes com início tardio, ocorrendo, em alguns estudos, em mais da metade dos olhos examinados21. Em casos isolados, podem até representar a primeira manifestação clínica detectável22. No entanto, estudos recentes indicam que, mesmo entre pacientes com sintomas neurológicos estabelecidos, as opacidades vítreas podem estar ausentes na avaliação inicial, sugerindo que o envolvimento ocular pode ser subestimado e surgir em uma fase posterior22.
Características clínicas
Observou-se que essas opacidades se formam no vítreo periférico e se estendem de forma centrípeta23. As fibras de colágeno do vítreo atuam como suporte para a agregação da TTR mutante, essa proteína apresenta um alto grau de afinidade com as membranas basais e com a matriz vítrea. Esta composta principalmente por colágeno tipo II e é estrutural e bioquimicamente semelhante ao colágeno das membranas basais, o que favorece a deposição amiloide. Dessa forma, as opacidades vítreas são caracterizadas por uma aparência clássica, descritas como semelhantes a folhas, películas, faixas, teias de aranha, algodão, fibras finas ou lã de vidro, sendo a última mais frequentemente relacionada à doença6.
As opacidades vítreas são causadas por debris celulares, proteínas, sangue, depósitos ou exsudatos inflamatórios suspensos no humor vítreo, cada causa apresenta diferentes padrões que são importantes para a sua identificação clínica. Destaca-se a sinérese vítrea que possui filamentos transparentes, móveis e flutuantes com o movimento ocular; a uveíte contém opacidades mais densas que reduzem a acuidade visual; cita-se, também, a hemorragia vítrea que se apresenta com um vermelho escuro característico24. Outra opacidade vítrea comumente encontrada, porém majoritariamente benigna e assintomática, é a hialose asteroide que possui pontos brancos amarelados e brilhantes aderidos à matriz vítrea25,26. O ultrassom ocular é importante ferramenta diagnóstica nesses casos27.
Na vATTR, a coriorretinopatia tem sido descrita como potencial manifestação ocular, frequentemente envolvendo alterações vasculares, tais como atenuação vascular da coroide ou oclusão vascular da coroide28. No entanto, neste paciente — apesar da doença avançada e da deposição proeminente de amiloide no vítreo — não foram observadas alterações vasculares no exame fundoscópico ou angiográfico. Esta ausência de envolvimento vascular da coroide sugere que a deposição amiloide ocular possa progredir para uma deficiência estrutural grave sem necessariamente induzir patologia vascular evidente; sugere, também, que tais alterações podem ser subclínicas, dificultando a detecção diagnóstica atual29. Além disso, o acompanhamento a longo prazo sugere que a função visual pode estar intacta nos primeiros anos após a detecção e que a deterioração visual pode levar vários anos29. Portanto, a avaliação multimodal é recomendada mesmo na ausência de alteração fundoscópicas iniciais.
Tratamento cirúrgico
O tratamento padrão para esta condição é a VVPP com peeling da membrana limitante interna, mas as opacidades podem apresentar recidiva devido à dispersão de depósitos residuais ou à produção intraocular contínua de TTR mutante30-32. Embora a VVPP melhore significativamente a acuidade visual, a recorrência das opacidades, a presença de neuropatia sensório-motora em agravamento, a disfunção autonômica grave e complicações intraoperatórias representam desafios no tratamento a longo prazo desses pacientes33-35. Neste caso, a ausência de recorrência após seis meses sugere provável resultado da remoção completa de todas as partes vítreas durante a VVPP. No entanto, ainda é possível ocorrer uma recorrência tardia devido à síntese EPR da TTR mutante6.
Após a cirurgia, esse paciente apresentou uma rápida evolução da catarata, o que também é esperado após a VVPP, dado que a formação de catarata representa a complicação mais comum da VVPP. A incidência de cirurgia de catarata após VVPP é estimada em aproximadamente 40% e pode aumentar para até 85% em cinco anos36,37. Frente a isso, a técnica combinada de VVPP com facoemulsificação em um único procedimento tem sido cada vez mais adotada com segurança38.
Terapias futuras
A terapia gênica é uma estratégia terapêutica futura promissora para a amiloidose por TTR, concentrando-se principalmente em estratégias para suprimir a expressão do gene TTR variante por meio da degradação do RNA mensageiro do TTR39,40. O tafamidis - uma das terapias anti-amiloides - tem como mecanismo de ação a estabilização da TTR mutante através da ligação aos sítios de ligação do T4 e estabilização do tetrâmero da TTR, permitindo a prevenção da dissociação em monômeros e intermediários amiloidógenos e tóxicos39. As indicações de uso dependem do estágio da doença, da idade do paciente, de doenças associadas, da gravidade da cardiopatia e das contraindicações. O tafamidis é preferencial para o tratamento do estágio inicial da PAF sintomática. A neuropatia deve ser sintomática, com mutação amiloidogênica da TTR, amiloidose comprovada por biópsia e estágio I da doença. Em estudo duplo cego randomizado, apresentou ausência de progressão da neuropatia em 60% dos pacientes no grupo tafamidis versus 38% no grupo placebo39.
Outra opção terapêutica é o patisiran (ALN-TTR02), que consiste em um siRNA específico para mRNA de TTR formulado em LNPs, com redução robusta, dose-dependente e estatisticamente significativa dos níveis séricos da proteína TTR40,41. Conforme estudo APOLLO42, o patisiran pode melhorar as manifestações neurológicas em receptores de terapia com dois fármacos (DLT) com AAP, sem produzir efeitos adversos relevantes. Além disso, outra medicação disponível é o inotersen, que foi recentemente aprovada pela FDA dos Estados Unidos para o tratamento da polineuropatia associada à amiloidose. Ao atingir os transcritos de mRNA da TTR, o inotersen diminui a tradução e a produção de proteínas, reduzindo o depósito de proteínas mal dobradas. O inotersen e o patisiran demonstraram excelentes resultados em pacientes com hATTR com polineuropatia. Tanto o inotersen quanto o patisiran retardaram a progressão da doença e até mesmo potencialmente reverteram a progressão43.
Análise histológica
O vermelho Congo sob luz polarizada é o principal método histológico utilizado para a identificação de depósitos amiloides44. Entretanto, devido a grande concentração de depósitos neste paciente, a luz direta foi suficiente para identificação do material. Embora não determine com precisão a natureza amiloide dos agregados proteicos, continua a ser o método mais rápido e prático para a coloração de amiloides in vitro44,45. Neste caso, o paciente apresentou um início tardio da doença, exigindo a exclusão de etiologias alternativas para as opacidades vítreas. O tratamento cirúrgico não só garantiu o prognóstico funcional, como a histologia foi essencial para confirmar a hipótese diagnóstica e excluir outras causas de opacidade vítrea.
No entanto, outros corantes também podem ser utilizados para a detecção desses depósitos. A tioflavina T é um corante fluorescente que se liga às fibrilas amiloides; após a ligação, a sua fluorescência aumenta significativamente, emitindo uma luz esverdeada que pode ser visualizada ao microscópio de fluorescência. Outro corante que pode ajudar na sua identificação é a hematoxilina e eosina (H&E). Embora a H&E não seja específica para amiloides, é a coloração mais amplamente utilizada na histopatologia geral — as proteínas amiloides aparecem, normalmente, como depósitos homogêneos de cor rosa na lâmina. Esta técnica não é conclusiva por si só e, portanto, não é recomendada como método principal para a identificação de amiloides44,46.
Este caso ilustra os desafios do tratamento oftalmológico em pacientes com doenças sistêmicas raras. O vítreo pode ser um local importante para a deposição amiloide, muitas vezes levando à deficiência visual progressiva, necessitando de tratamento cirúrgico precoce. A VVPP, quando indicada adequadamente, continua a ser o principal tratamento, proporcionando tanto a confirmação diagnóstica como a recuperação funcional. Em última análise, o acompanhamento multidisciplinar é essencial, pois a avaliação oftalmológica precoce e a intervenção oportuna podem preservar a função visual e melhorar a qualidade de vida geral de indivíduos com amiloidose sistêmica. Este caso destaca a importância da avaliação oftalmológica precoce em pacientes com amiloidose transtirretina hereditária. A amiloidose vítrea pode ser o achado ocular inicial ou predominante, e a VVPP permite tanto a confirmação diagnóstica quanto uma recuperação visual. O acompanhamento abrangente e multidisciplinar é essencial no tratamento das manifestações oculares e sistêmicas da ATTRv.
AGRADECIMENTOS
Este estudo foi apoiado por Fundação de Amparo à Pesquisa do Estado de São Paulo, pelo autor LES (número 2024/16042-9).
REFERÊNCIAS
1. Sipe JD, Cohen AS. Review: History of the Amyloid Fibril. J Struct Biol. 2000;130(2-3):88-98.
2. Cohen AS, Calkins E. Electron Microscopic Observations on a Fibrous Component in Amyloid of Diverse Origins. Nature. 1959;183(4669):1202-3.
3. Vaxman I, Gertz M. When to Suspect a Diagnosis of Amyloidosis. Acta Haematol. 2020;143(4):304-311.
4. Blancas-Mejía LM, Ramirez-Alvarado M. Systemic Amyloidoses. Annu Rev Biochem. 2013 Jun 2;82(1):745-74.
5. Comenzo RL. Amyloidosis. Curr Treat Options Oncol. 2006;7(3):225-36.
6. Minnella AM, Rissotto R, Antoniazzi E, Di Girolamo M, Luigetti M, Maceroni M, et al. Ocular Involvement in Hereditary Amyloidosis. Genes (Basel). 2021;12(7):955.
7. Carroll A, Dyck PJ, de Carvalho M, Kennerson M, Reilly MM, Kiernan MC, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2022;93(6):668-678.
8. Munar-Qués M, Salvá-Ladaria L, Mulet-perera P, Solé M, López-Andreu FR, Saraiva MJM. Vitreous amyloidosis after liver transplantation in patients with familial amyloid polyneuropathy: Ocular synthesis of mutant transthyretin. Amyloid. 2000;7(4):266-9.
9. Kiuru-Enari S, Haltia M. Hereditary gelsolin amyloidosis. Handb Clin Neurol. 2013;115:659-81.
10. Kawaji T, Ando Y, Ando E, Nakamura M, Hirata A, Tanihara H. A case of vitreous amyloidosis without systemic symptoms in familial amyloidotic polyneuropathy. Amyloid. 2004;11(4):257-9.
11. Salvador F, Mateo C, Alegre J, Reventos A, García-Arumi J, Corcostegui B. Vitreous amyloidosis without systemic or familial involvement. Int Ophthalmol. 1993;17(6):355-7.
12. Togashi S, Watanabe H, Nagasaka T, Shindo K, Shiozawa Z, Maeda S, et al. An aggressive familial amyloidotic polyneuropathy caused by a new variant transthyretin Lys 54. Neurology. 1999;53(3):637-9.
13. Mazzeo A, Russo M, Di Bella G, Minutoli F, Stancanelli C, Gentile L, et al. Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area. J Neuromuscul Dis. 2015;2(s2):S39-S48.
14. Ueno S, Uemichi T, Yorifuji S, Tarui S. A novel variant of transthyretin (Tyr114 to Cys) deduced from the nucleotide sequences of gene fragments from familial amyloidotic polyneuropathy in Japanese sibling cases. Biochem Biophys Res Commun. 1990;169(1):143-7.
15. Levy J, Hawkins PN, Rowczenio D, Godfrey T, Stawell R, Zamir E. Familial amyloid polyneuropathy associated with the novel transthyretin variant Arg34Gly. Amyloid. 2012;19(4):201-3.
16. Martin SE, Benson MD, Hattab EM. The pathologic spectrum of oculoleptomeningeal amyloidosis with Val30Gly transthyretin gene mutation in a postmortem case. Hum Pathol. 2014;45(5):1105-8.
17. Waddington-Cruz M, Wixner J, Amass L, Kiszko J, Chapman D, Ando Y; THAOS investigators. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. 2021;10(2):753-66.
18. Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63(1):129-38.
19. Correia AS, Mendonça M, Caetano A, Medeiros E. A sporadic case of late-onset familial amyloid polyneuropathy with a monoclonal gammopathy. Neuromuscul Disord. 2015;25(8):658-60.
20. Obi CA, Mostertz WC, Griffin JM, Judge DP. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist Debakey Cardiovasc J. 2022;18(2):17-26.
21. Ferreira N, Dias D, Coelho T. Specific ophtalmologic changes in late onset familial amyloid polyneuropathy (FAP) portuguese patients. Orphanet J Rare Dis. 2015 Nov 2;10(Suppl 1):P63.
22. Gondim FAA, Holanda Filha JG, Moraes Filho MO. Ophthalmological manifestations of hereditary transthyretin amyloidosis. Arq Bras Oftalmol. 2022;85(5):528-538.
23. Mathew R, Zeppieri M. Ocular Amyloidosis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025 Jan. 2024 May 28.
24. Milston R, Madigan MC, Sebag J. Vitreous floaters: Etiology, diagnostics, and management. Surv Ophthalmol. 2016;61(2):211-27.
25. Zaher E, Blumenthal Y, Blumenthal EZ. The lack of floater perception in eyes with asteroid hyalosis and its direct implications on laser vitreolysis. Int J Retina Vitreous. 2024;10(1):80.
26. Snehi S, Bang PH, Mamtani N, Singh K. Galactic vision: asteroid hyalosis in vitreous with moonlit optic disc. BMJ Case Rep. 2024;17(5):e260387.
27. Budhram G, Cronsell J, Schroeder M, Sautner J, Schoenfeld E, Elia T, et al. Mobile vitreous opacities on ocular ultrasonography are not always pathologic: a cross-sectional survey in an asymptomatic population. Am J Emerg Med. 2015;33(12):1808-13.
28. Roybal CN, Sanfilippo CJ, Nazari H, Law JC, Bhaleeya S, Chui Ming GC, et al. multimodal imaging of the retina and choroid in systemic amyloidosis. Retin Cases Brief Rep. 2015;9(4):339-46.
29. Nagura K, Inoue T, Ching J, Sato A, Kitahata S, Maruyama-Inoue M, et al. Long-term follow-up of a case of amyloidosis-associated chorioretinopathy. Am J Ophthalmol Case Rep. 2020 Sep 2;19:100846.
30. Venkatesh P, Selvan H, Singh SB, Gupta D, Kashyap S, Temkar S, et al. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017;124(7):1014-1022.
31. Ferreira NN, Dias DAC, Carvalho RPA, Coelho MTPM. Re-intervention in de novo vitreous opacities after pars plana vitrectomy in familial amyloidotic polyneuropathy TTR VAL30METPORTUGUESE PATIENTS. Retin Cases Brief Rep. 13(3):273-8.
32. Beirão NM, Matos E, Beirão I, Costa PP, Torres P. Recurrence of vitreous amyloidosis and need of surgical reintervention in Portuguese patients with familial amyloidosis ATTR V30M. Retina. 2011;31(7):1373-7.
33. Xu Q, Wang X, Zhang Z, Li J, Liu H, Ji S, et al. Case Report: Vitreous Amyloidosis Caused by a TTR Lys55Asn Mutation With Intraoperative Suprachoroidal Hemorrhage. Front Med (Lausanne). 2022 Jan 24;8:797223.
34. Xue YW, Xiao YQ. Vitreous amyloidosis caused by Lys55Asn mutation in TTR with peripheral neuropathy onset: a case report of FAP-related complications. Int J Ophthalmol. 2025;18(1):184-186.
35. Tan Y, Tao Y, Sheng YJ, Zhang CM. Vitreous amyloidosis caused by a Lys55Asn variant in transthyretin: A case report. World J Clin Cases. 2022;10(32):12000-12006.
36. Jackson TL, Donachie PHJ, Sparrow JM, Johnston RL. United Kingdom National Ophthalmology Database Study of Vitreoretinal Surgery: report 1; case mix, complications, and cataract. Eye (Lond). 2013;27(5):644-51.
37. Soliman MK, Hardin JS, Jawed F, Uwaydat SH, Faramawi MF, Chu CJ, et al. A Database Study of Visual Outcomes and Intraoperative Complications of Postvitrectomy Cataract Surgery. Ophthalmology. 2018;125(11):1683-91.
38. Farahvash A, Popovic MM, Eshtiaghi A, Kertes PJ, Muni RH. Combined versus Sequential Phacoemulsification and Pars Plana Vitrectomy: A Meta-Analysis. Ophthalmol Retina. 2021;5(11):1125-1138.
39. Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6(2):129-39.
40. Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086-97.
41. Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015 Dec 4;10:109.
42. Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21.
43. Robinson C, Pham C, Zamarripa AM, Dugay CS, Lee CA, Berger AA, et al. Inotersen to Treat Polyneuropathy Associated with Hereditary Transthyretin (hATTR) Amyloidosis. Health Psychol Res. 2022;10(5):67910.
44. Yakupova EI, Bobyleva LG, Vikhlyantsev IM, Bobylev AG. Congo Red and amyloids: history and relationship. Biosci Rep. 2019;39(1):BSR20181415.
45. Howie AJ, Brewer DB, Howell D, Jones AP. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab Invest. 2008;88(3):232-42.
46. Xue C, Lin TY, Chang D, Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R Soc Open Sci. 2017;4(1):160696.
Financiamento: Declaram não haver.
Aprovado pelo seguinte Comitê de Ética: Universidade Positivo (número 5.504.706).
Conflitos de interesse: Declaram não haver.
Recebido em:
3 de Novembro de 2025.
Aceito em:
16 de Janeiro de 2026.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados









 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket