

Marcia Beatriz Tartarella1,2; Islane Verçosa1; Renata Girão Cavalcante1; Paula Carneiro1; Paloma Verçosa1; João Borges Fortes Filho4; Erlane Marques Ribeiro3
DOI: 10.17545/eOftalmo/2023.0048
RESUMO
Hipoplasia dérmica focal ou síndrome de Goltz, é uma doença genética caracterizada por anomalias oculares e envolvimento multissistêmico devido à displasia dos tecidos derivados do mesoectoderma, A avaliação oftalmológica foi realizada em duas pacientes com diagnóstico clínico de Síndrome de Goltz com anomalias em pele, nos dentes e esqueléticas, que apresentaram as seguintes alterações oculares: microftalmia, catarata, coloboma parcial de íris, coloboma de retina, descolamento de retina e alterações palpebrais.
Palavras-chave: Síndrome de Goltz; Hipoplasia dérmica focal; microftalmia; Íris coloboma; Catarata congênita.
ABSTRACT
Focal dermal hypoplasia, or Goltz syndrome, is a genetic disorder characterized by ocular and systemic abnormalities due to dysplasia of mesoectodermal-derived tissues. This study describes the ophthalmological features of two patients with Goltz syndrome presenting ocular, cutaneous, dental, and skeletal anomalies. Ocular manifestations included microphthalmia, cataracts, retinal coloboma, retinal detachment, iris coloboma, and eyelid abnormalities.
Keywords: Focal dermal hypoplasia; Goltz syndrome; Iris coloboma; Microphthalmia; Pediatric cataract.
INTRODUÇÃO
A síndrome de Goltz (SG) é uma anomalia genética rara. Sua incidência é desconhecida e poucos casos foram descritos na literatura. A prevalência estimada de SG pode variar de 1/30.827 até 1/256.000. É causada por mutação no gene POCRN, com herança dominante ligada ao X e apresenta hipoplasia dérmica focal (HDF).
Ocorre uma alteração no desenvolvimento do mesoectoderma que é caracterizada por alterações em múltiplos sistemas Ocorrem anomalias cutâneas, oculares, dentárias, esqueléticas, no trato urinário, gastrointestinal, cardiovascular e no sistema nervoso central. Pacientes com SG podem apresentar uma facies típica, com queixo pontiagudo, assimetria facial e alae nasi alargada1,2.
As manifestações oftalmológicas descritas na SG incluem anoftalmia, microftalmia, ectrópio palpebral, hipertelorismo, coloboma, aniridia, catarata congênita, alterações pigmentares retinianas, papilomas da conjuntiva e pálpebra, anormalidades no sistema lacrimal. Estas alterações oftalmológicas podem ocorrer em 40% a 77% dos casos1,2.
O presente artigo relata as alterações oftalmológicas em dois pacientes com diagnóstico de síndrome de Goltz.
RELATO DE CASOS
Caso 1
Paciente, do sexo feminino, 6 anos de idade, avaliada e diagnosticada como SG pelo geneticista e pelo clínico, foi encaminhada ao Hospital de Olhos CAVIVER (Centro de Aperfeiçoamento Visual Ver a Esperança Renascer, Fortaleza, CE, Brasil) para avaliação oftalmológica.
A paciente apresentava assimetria facial com formato triangular (Figura 1A), lesões atróficas lineares de pele seguindo as linhas de Blaschko; alopecia focal (Figura 1B); anomalia de implantação dos polegares com deformidade das mãos em "garra de lagosta" (Figura 1C), baixa implantação das orelhas, hipertelorismo e anomalias dentárias e orofaciais. A história familiar foi negativa para SG.
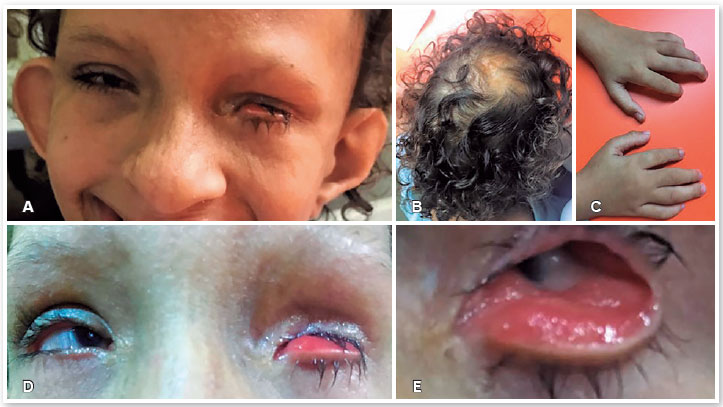
Ao exame oftalmológico apresentou acuidade visual de 20/400 no olho direito (OD) e percepção luminosa no olho esquerdo (OE). Microftalmia e microcórnea estavam presentes em ambos os olhos. No OD observou-se córnea transparente com coloboma inferior parcial de íris e catarata subcapsular posterior. No OE detectou-se leucoma envolvendo toda a córnea. O diâmetro horizontal da córnea obtido foi de 6mm no OD e 3mm no OE. O OE apresentava ectrópio da pálpebra inferior (Figuras 1D e 1E), com sintomatologia de lacrimejamento e olho vermelho, sendo encaminhada ao serviço de plástica ocular para avaliação e tratamento do ectrópio, e para analise de uma possível correção estética neste olho.
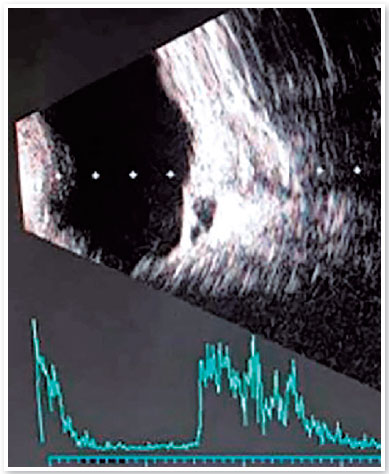
A medida do diâmetro axial de OD foi 19mm, e no OE foi impossibilitada a aferição devido extrema microftalmia. A avaliação da paquimetria apresentou espessura média da córnea central de 680 micra em OD e 580 micra em OE. O exame do fundo de olho revelou um coloboma inferior de retina se estendendo até o nervo óptico em OD. O exame de ultrassonografia no OD revelou um cisto de escavação e, no OE, um descolamento seroso total da retina (Figura 2). A acuidade visual obtida em OD foi de 20/40 com a correção da refração prescrita em óculos de -5,00 esf = -3,00 cil (40º).
Caso 2
Paciente, sexo feminino, de 17 anos, foi encaminhada ao Hospital de olhos CAVIVER. A avaliação clínica, com quadro característico, e a avaliação genética evidenciaram o diagnóstico de SG aos cinco anos de idade.
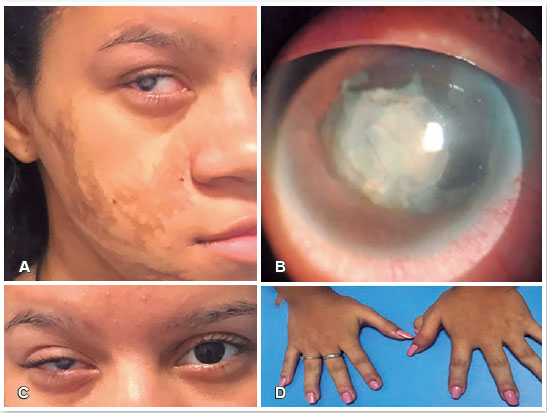
A paciente apresentava lesões hiperpigmentadas lineares e atróficas na pele na hemiface direita (Figura 3A), anormalidades dentárias, polidactilia em mão esquerda (Figura 3D) e leucocoria em OD.
Ao exame oftalmológico apresentou acuidade visual em OD sem percepção luminosa e 20/60 em OE. A paciente obteve visão de 20/20 em OE com prescrição de óculos de -1.00 dioptria esférica.
Na avaliação de segmento anterior do OD foi detectada microcórnea com diâmetro horizontal de 8 mm, coloboma de íris na região nasal, leucocoria secundária a presença de catarata membranosa e vascularizada, e sinéquias posteriores (Figuras 3B e 3C). A avaliação do segmento anterior do OE estava normal, com cristalino e córnea transparentes, e apresentando diâmetro corneano de 11 mm. Ecografia realizada em OD estava normal e a avaliação de fundo de olho em OE estava normal.
Optou-se por prescrever uma lente de contato cosmética colorida com o objetivo de melhorar a estética ocular devido a presença de leucocoria no OD.
DISCUSSÃO
A Hipoplasia dérmica focal ou Síndrome de Goltz é uma anomalia genética multissistêmica1,3,4. As manifestações oftalmológicas mais frequentes na HDF descritas na literatura são os colobomas e a microftalmia. Outros achados oculares reportados incluem anoftalmia, catarata, hipertelorismo, ectrópio palpebral, ptose palpebral, obstrução de ducto nasolacrimal, papilomas conjuntivais ou da margem palpebral, opacidades corneanas, aniridia, heterocromia, opacidades vítreas, e, hipoplasia de nervo óptico2,4. Poucos casos de HDF unilateral foram previamente publicados5.
O diagnóstico diferencial incluía a síndrome de microftalmia, incontinentia pigmenti, aplasia dermal e esclerocornea (MIDAS). Porém a incontinentia pigmenti apresenta lesões cutâneas verrucosas e com vesículas com hiperpigmentação, que diferem das áreas lineares atróficas da HDF2.
Os casos apresentados aqui manifestaram características clínicas típicas de SG e apresentaram as seguintes manifestações oculares: microftalmia, microcornea, catarata, coloboma parcial de íris, coloboma de retina, descolamento de retina e ectropia palpebral.
O tratamento dos pacientes com SG geralmente é paliativo e necessita de uma equipe multidisciplinar. A conduta oftalmológica pode incluir procedimentos estéticos para microftalmia ou para leucocoria, cirurgia para correção de ectrópio palpebral, e, cirurgia de catarata quando necessário. A conduta, tanto funcional quanto estética, foi planejada para garantir uma melhor qualidade de vida para ambos os pacientes. Nos dois casos obteve-se melhor acuidade visual com a prescrição de lentes corretivas.
No caso 2, o OD apresentava microftalmia e catarata membranosa vascularizada ocasionando uma leucocoria, então, uma lente de contato cosmética colorida foi adaptada.
Catarata não é a anomalia mais frequente em SG, podendo ocorrer de 11% a 17 % dos pacientes1,2. Ambas as pacientes deste estudo apresentaram catarata. No caso 1 a catarata era subcapsular posterior, e no caso 2 o cristalino estava em reabsorção apresentando uma catarata membranosa vascularizada, provavelmente devido a uma opacificação progressiva do cristalino. Este fato demonstra a importância de uma avaliação oftalmológica precoce e tratamento adequado em pacientes com SG para evitar ambliopia irreversível ou deficiências visuais.
A análise do estado refracional dos pacientes e prescrição de óculos foi essencial para maximizar o potencial de visão em ambos os casos. É importante prescrever lentes protetivas para prevenir acidentes oculares em crianças com visão monocular. Ambas as pacientes obtiveram melhora da visão em pelo menos um olho.
A síndrome de Goltz apresenta uma alta prevalência de anomalias oculares, sendo de extrema importância encaminhar os pacientes para avaliação oftalmológica. Os dois casos aqui descritos apresentaram microftalmia, catarata e déficits visuais. Os procedimentos oftalmológicos adotados incluíram o tratamento estético da microftalmia, da leucocoria e do ectrópio palpebral; e a prescrição de óculos. Portanto, um diagnóstico e tratamento precoces das condições oculares são de grande importância para se obter o potencial visual máximo de cada paciente e uma melhor qualidade de vida.
REFERÊNCIAS
1. Gisseman JD, Herce HH. Ophthalmologic manifestations of Focal Dermal Hypoplasia (Goltz syndrome): A case series of 18 patients. Am J Med Genet C Semin Med Genet. 2016; 172C(1):59-63.
2. Moramarco A, Himmelblau E, Miraglia E, Mallone F, Robert V, Franzone F, et al. Ocular manifestations in Gorlin-Goltz syndrome. Orphanet J Rare Dis. 2019;14(1):218.
3. Harmsen MB, Azzarello-Burri S, García González MM, Gillessen-Kaesbach G, Meinecke P, Müller D et al. Goltz-Gorlin (focal dermal hypoplasia) and the microphthalmia with linear skin defects (MLS) syndrome: no evidence of genetic overlap. Eur J Hum Genet. 2009;17(10):1207-15.
4. Souza-e-Souza I, Cunha PCAS. Síndrome de Goltz: relato de dois casos. An Bras Dermatol. 2003;78(1):91-7.
5. Tenkir A, Teshome S. Goltz syndrome (focal dermal hypoplasia) with unilateral ocular, cutaneous and skeletal features: case report. BMC Ophthalmol. 2010 Nov19:10:28.
| INFORMAÇÃO DOS AUTORES |
|
 |
» Marcia Beatriz Tartarella http://orcid.org/0000-0003-2361-3355 http://lattes.cnpq.br/6983200921618896 |
 |
» Islane Maria Castro Verçosa http://orcid.org/0000-0002-6669-7934 http://lattes.cnpq.br/8594289814981440 |
 |
» Paloma Verçosa http://orcid.org/0000-0002-3590-3459 http://lattes.cnpq.br/8162696890492848 |
 |
» Erlane Marques Ribeiro http://orcid.org/0000-0002-7104-0128 http://lattes.cnpq.br/3638959901261806 |
 |
» Renata Girão Cavalcante http://orcid.org/0000-0002-3853-9429 http://lattes.cnpq.br/9176734716498992 |
 |
» Paula Carneiro http://orcid.org/0000-0001-7649-1983 http://lattes.cnpq.br/2188797319991075 |
 |
» João Borges Fortes Filho http://orcid.org/0000-0001-5682-0962 http://lattes.cnpq.br/0510091397232374 |
Financiamento: Declaram não haver.
Aprovado pelo seguinte comitê de ética em pesquisa: Hospital Infantil Albert Sabin (CAAE: 78568717.0.0000.5042).
Conflitos de Interesse: Declaram não haver.
Recebido em:
6 de Setembro de 2023.
Aceito em:
11 de Outubro de 2023.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket