

Marcia Beatriz Tartarella1,2; Islane Verçosa1; Renata Girão Cavalcante1; Paula Carneiro1; Paloma Verçosa1; João Borges Fortes Filho4; Erlane Marques Ribeiro3
DOI: 10.17545/eOftalmo/2023.0048
ABSTRACT
Focal dermal hypoplasia, or Goltz syndrome, is a genetic disorder characterized by ocular and systemic abnormalities due to dysplasia of mesoectodermal-derived tissues. This study describes the ophthalmological features of two patients with Goltz syndrome presenting ocular, cutaneous, dental, and skeletal anomalies. Ocular manifestations included microphthalmia, cataracts, retinal coloboma, retinal detachment, iris coloboma, and eyelid abnormalities.
Keywords: Focal dermal hypoplasia; Goltz syndrome; Iris coloboma; Microphthalmia; Pediatric cataract.
RESUMO
Hipoplasia dérmica focal ou síndrome de Goltz, é uma doença genética caracterizada por anomalias oculares e envolvimento multissistêmico devido à displasia dos tecidos derivados do mesoectoderma, A avaliação oftalmológica foi realizada em duas pacientes com diagnóstico clínico de Síndrome de Goltz com anomalias em pele, nos dentes e esqueléticas, que apresentaram as seguintes alterações oculares: microftalmia, catarata, coloboma parcial de íris, coloboma de retina, descolamento de retina e alterações palpebrais.
Palavras-chave: Síndrome de Goltz; Hipoplasia dérmica focal; microftalmia; Íris coloboma; Catarata congênita.
INTRODUCTION
Goltz syndrome (GS) is a rare genetic anomaly. The incidence of GS is unknown, and only a few cases have been described in the literature. The estimated prevalence is 1/30,827-1/256,000, caused by a mutation in the POCRN gene with X-linked dominant inheritance. The primary manifestation is focal dermal hypoplasia (FDH). There is abnormal mesoectodermal development, and the syndrome is clinically characterized by skin, eyes, teeth, skeleton, and urinary anomalies, as well as the gastrointestinal, cardiovascular, and central nervous systems. Most patients with GS show typical facies (facial asymmetry, pointed chin, and notched alae nasi)1,2. The ocular GS manifestations include anophthalmia, microphthalmia, eyelid ectropion, hypertelorism, coloboma, aniridia, congenital cataracts, retinal pigment changes, conjunctiva and eyelid papillomas, and lacrimal system abnormalities. Ophthalmic manifestations occur in 40%-77% of cases1,2.
The present study reports the ophthalmological features and management of two patients with GS.
CASE REPORT
Case 1
A 6-year-old woman with an existing diagnosis of GS based on genetic and clinical evaluations was referred to the CAVIVER Eye Clinic (Centro de Aperfeiçoamento Visual Ver a Esperança Renascer, Fortaleza, CE, Brazil) for an ophthalmic evaluation. The patient presented with asymmetric triangular facies (Figure 1A), atrophic linear skin lesions following Blaschko lines, focal alopecia (Figure 1B), lobster claw deformity of the hands (Figure 1C), low ear implantation, hypertelorism, and dental and orofacial abnormalities, with no family history of GS.
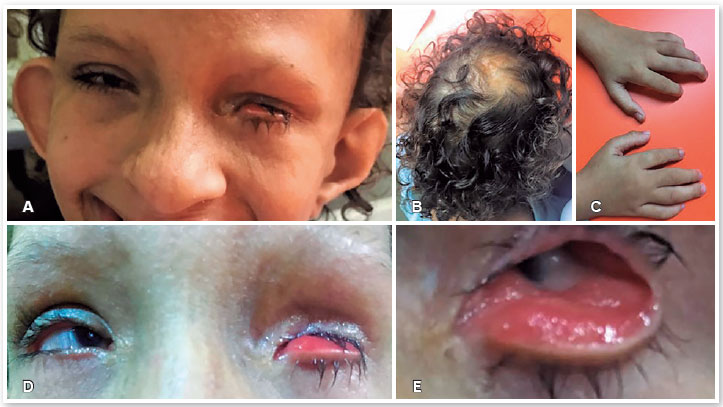
The patient's visual acuity in the right eye (RE) was 20/400, whereas the patient had no light perception in the left eye (LE). Microphthalmia and microcornea were noted in both eyes. RE had a small clear cornea. A partial iris coloboma and a posterior subcapsular cataract were observed. In LE, there was a leukoma affecting the entire cornea. The horizontal diameters of the corneas were 6 mm and 3mm in the RE and LE. LE presented inferior eyelid ectropion (Figures 1D and 1E), with the symptomatology of tearing and red eye. The patient was referred to our oculoplastic department for evaluation and treatment of the eyelid ectropion and planning aesthetic management.
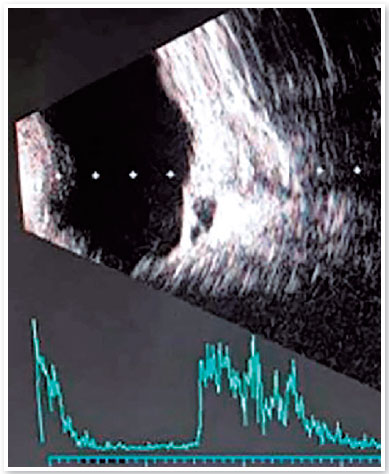
The RE axial length measurement was 19mm. The LE axial length could not be measured because of extreme microphthalmia. Pachymetry disclosed 680 and 580 micra corneal thickness in RE and LE. An eye fundus evaluation of RE showed an inferior coloboma of the retina extending to the optic nerve. Ocular ultrasonography disclosed a choroidal excavation cyst in RE, and a total serous retinal detachment in LE (Figure 2). The best corrected visual acuity achieved in RE was 20/40, with a prescription of -5.00 -3.00 cyl 40º glasses.
Case 2
A 17-year-old woman was referred to the CAVIVER Eye Clinic. Genetic and clinical evaluations of the patient at the age of 5 resulted in the GS diagnosis. She presented asymmetric hyperpigmented atrophic linear skin lesions on the right side of her face (Figure 3A) and dental abnormalities, RE leukocoria, and polydactyly of the left hand (Figure 3D).
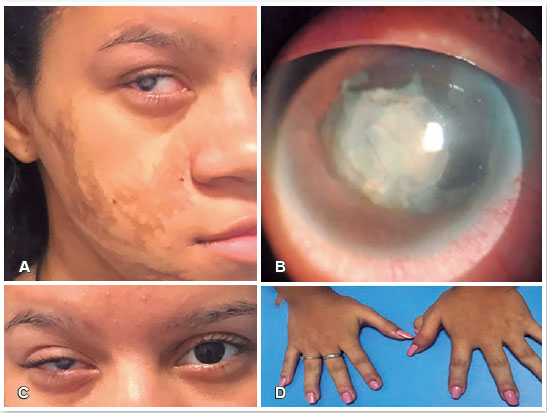
Ophthalmic evaluation found no light perception in RE and visual acuity of 20/60 in LE. The patient achieved the best corrected visual acuity of 20/20 in LE with the prescription of a -1.00-diopter spherical lens. Evaluation of the anterior segment in RE detected microcornea with a horizontal corneal diameter of 8mm, nasal iris coloboma, leukocoria secondary to a dense, vascularized, membranous lens, and posterior synechiae (Figures 3C and 3B). The anterior segment in LE was normal, with a clear lens and cornea. The horizontal corneal diameter of LE was 11mm. Echography was normal in RE. Eye fundus evaluation was normal in LE. Due to leukocoria in RE, cosmetic tinted contact lenses were prescribed to improve ocular aesthetics.
DISCUSSION
Focal dermal hypoplasia, or Goltz Syndrome,, is a genetic multisystemic disorder1,3,4. The most frequent ophthalmic manifestations described in the literature are colobomas and microphthalmia. Reportedly, other ocular findings include anophthalmia, cataracts, hypertelorism, eyelid ectropion, ptosis, nasolacrimal duct obstruction, lid margin or conjunctival papillomas, corneal opacities, aniridia, heterochromia, cloudiness of the vitreous humor, and optic nerve hypoplasia2-4. To date, only a few cases of unilateral FDH have been published5.
The differential diagnosis of GS includes microphthalmia, incontinentia pigmenti, dermal aplasia, and sclerocornea (MIDAS syndrome). A clinical history of incontinentia pigmenti presents cutaneous vesiculation and verrucous lesions with hyperpigmentation, helping differentiate it from FDH, which presents with linear atrophic areas2.
Both cases reported here had typical clinical features of GS and presented the following ocular manifestations: microphthalmia, microcornea, cataracts, partial iris coloboma, retinal coloboma, retinal detachment, and eyelid ectropium. Treating patients with GS primarily requires a multidisciplinary team. Ophthalmic treatments may include aesthetic management of microphthalmia, surgical correction of eyelid ectropium, and cataract surgery. With both the patients in this report, the ophthalmological management aimed to improve the quality of life. Both patients achieved better vision with their lens prescriptions.
In the second case, the microphthalmic RE showed a membranous and vascularized cataract and leukocoria, and a cosmetic tinted contact lens was a suitable choice. Cataracts are not the most frequent ocular anomaly, occurring in 11%-17% of patients with GS1,2; however, both patients in this study presented it. The patient in Case 1 had a posterior subcapsular cataract, and the patient in Case 2 presented a reabsorbed lens and membranous vascularized cataract, probably due to the progressive opacification of the lens. This highlights the importance of early ophthalmic assessment and prompt treatment in patients with GS to avoid visual impairment.
Determination of the refractive status and eyeglass prescription helped improve visual acuity in both cases. Prescribing glasses with protective lenses to prevent eye injuries in children with monocular vision is important. Both patients achieved better vision in at least one eye.
There is a high prevalence of severe ocular abnormalities in patients with GS and referring all GS patients for comprehensive ophthalmic evaluations is vital. The two cases reported in this study presented microphthalmia, cataracts, and visual deficits. Ophthalmological assessments and treatments included aesthetic management of the microphthalmia, leukocoria, eyelid ectropium, and lens prescriptions. Therefore, early diagnosis and treatment of ocular conditions in patients with GS are essential to achieve maximum visual potential and optimize the patient's quality of life.
REFERENCES
1. Gisseman JD, Herce HH. Ophthalmologic manifestations of Focal Dermal Hypoplasia (Goltz syndrome): A case series of 18 patients. Am J Med Genet C Semin Med Genet. 2016; 172C(1):59-63.
2. Moramarco A, Himmelblau E, Miraglia E, Mallone F, Robert V, Franzone F, et al. Ocular manifestations in Gorlin-Goltz syndrome. Orphanet J Rare Dis. 2019;14(1):218.
3. Harmsen MB, Azzarello-Burri S, García González MM, Gillessen-Kaesbach G, Meinecke P, Müller D et al. Goltz-Gorlin (focal dermal hypoplasia) and the microphthalmia with linear skin defects (MLS) syndrome: no evidence of genetic overlap. Eur J Hum Genet. 2009;17(10):1207-15.
4. Souza-e-Souza I, Cunha PCAS. Síndrome de Goltz: relato de dois casos. An Bras Dermatol. 2003;78(1):91-7.
5. Tenkir A, Teshome S. Goltz syndrome (focal dermal hypoplasia) with unilateral ocular, cutaneous and skeletal features: case report. BMC Ophthalmol. 2010 Nov19:10:28.
| AUTHORS INFORMATIONS |
|
 |
» Marcia Beatriz Tartarella http://orcid.org/0000-0003-2361-3355 http://lattes.cnpq.br/6983200921618896 |
 |
» Islane Maria Castro Verçosa http://orcid.org/0000-0002-6669-7934 http://lattes.cnpq.br/8594289814981440 |
 |
» Paloma Verçosa http://orcid.org/0000-0002-3590-3459 http://lattes.cnpq.br/8162696890492848 |
 |
» Erlane Marques Ribeiro http://orcid.org/0000-0002-7104-0128 http://lattes.cnpq.br/3638959901261806 |
 |
» Renata Girão Cavalcante http://orcid.org/0000-0002-3853-9429 http://lattes.cnpq.br/9176734716498992 |
 |
» Paula Carneiro http://orcid.org/0000-0001-7649-1983 http://lattes.cnpq.br/2188797319991075 |
 |
» João Borges Fortes Filho http://orcid.org/0000-0001-5682-0962 http://lattes.cnpq.br/0510091397232374 |
Funding: No specific financial support was available for this study.
Approved by the following research ethics committee: Hospital Infantil Albert Sabin (CAAE: 78568717.0.0000.5042).
Conflict of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
September 6, 2023.
Accepted on:
October 11, 2023.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket