

Felipe Leão de Lima; Charles Porto Petruceli Carayon; Lara Lopes de Almeida
DOI: 10.17545/eOftalmo/2021.0032
ABSTRACT
Neurofibromatosis is an autosomal dominant disease with varying clinical presentations affecting several tissues and organs and may present with remarkable ophthalmological manifestation. This study presents the clinical case of an 8-year-old child diagnosed with type 1 neurofibromatosis during an elective ophthalmologic examination.
Keywords: Neurofibromatosis; Von Recklinghausen; Lisch nodules; NF1; café au lait macules; Glioma; Optic nerve.
RESUMO
A neurofibromatose é uma doença autossômica dominante com apresentação clínica variada acometendo diversos tecidos e órgãos, podendo se apresentar com rica manifestação oftalmológica. O presente trabalho apresenta o caso clínico de uma criança, 8 anos de idade, diagnosticada com neurofibromatose tipo 1 em exame oftalmológico eletivo.
Palavras-chave: neurofibromatose; Von Recklinghausen; Nódulos de Lisch; NF1; Manchas café-com-leite; Glioma; nervo óptico.
INTRODUCTION
Neurofibromatosis represents a spectrum of diseases of autosomal dominant origin. It has an incidence of approximately 1/3,000 live births1 and is characterized by insufficient synthesis of the protein neurofibromin-a tumor suppressor protein-and consequent disorder of the neurons, oligodendrocytes, astrocytes, leukocytes, and adrenal medulla2.
It mostly diagnosed clinically, and definitive diagnosis can be achieved by performing tumor biopsy3. It is essential that ophthalmologists have sufficient knowledge regarding this group of pathologies because they are often responsible for the initial diagnosis and early management of the condition. This is the report of a case of NF1 associated with optic nerve glioma diagnosed through important changes observed during ophthalmologic examination.
CASE REPORT
D.M.C, 8 years old, underwent elective ophthalmic consultation due to complaints of low visual acuity, mainly in the left eye, with insidious and progressive onset since approximately 6 months.
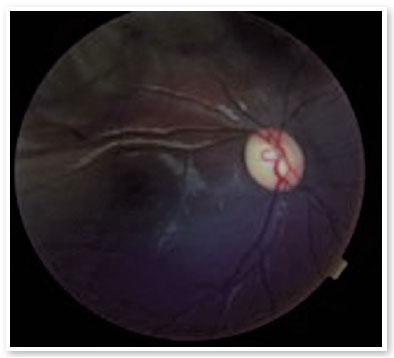
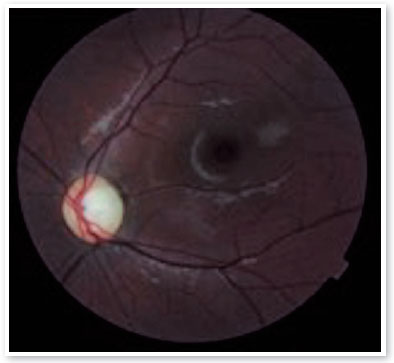
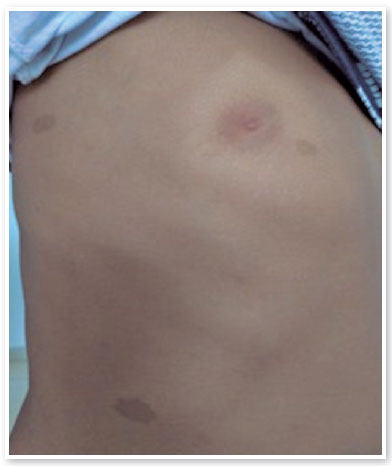
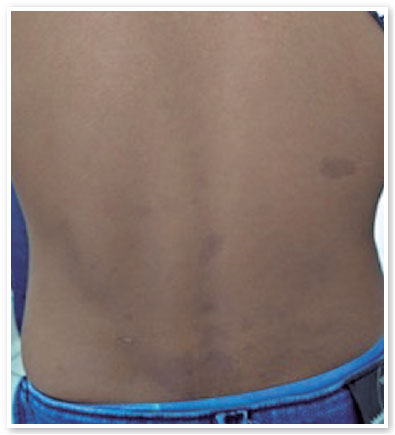
On ophthalmologic examination, static refraction of +1.50D spherical in both eyes (AO), with visual acuity of 20/20 in the right eye (RE), partial 20/100 in the left eye (LE) and changes in the Ishihara color test. There were no changes in the extrinsic ocular motility. On biomicroscopy, isochoric and photoreactive pupils were observed, with hamartomatous irian nodulations in AO, which were more concentrated inferiorly and suggestive of Lisch nodules (Figure 1). Gonioscopy demonstrated an open angle to the ciliary body in AO, and fundoscopy demonstrated a more pronounced bilateral optic papilla pallor in LE, with an excavation/disc ratio of 0.4 RE and 0.8 LE (Figures 2 and 3). The intraocular pressure was 12 mmHg in AO. On general inspection, the presence of numerous hyperchromic café au lait spots distributed on the trunk (Figure 4) and back (Figure 5) was noted.

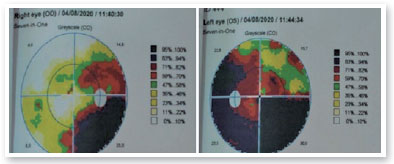
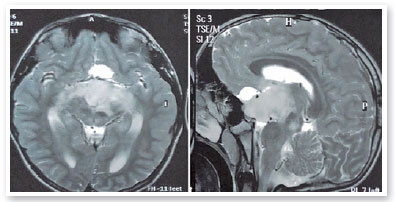
Complementary propaedeutics with 24-2 visual field test was requested; it showed, with good reliability, lower right quadrantopia in AO (Figure 6), which was observed to be deeper in LE. Brain MRI showed the presence of large, expansive, and invasive tissue formation at the base of the skull, with indefinite boundaries and irregular contours measuring approximately 3.9-4.7 cm in its largest orthogonal axes in the sagittal plane; involving the stem, pituitary infundibulum, optic nerves, and optic chiasm; and extending to the left nucleocapsular region and cerebral peduncle on the same side; these findings were highly suggestive of optic nerve glioma (Figure 7). Subsequently, we referred to the pediatric neurology and oncology service of Santa Casa de Belo Horizonte, and it was decided to perform a biopsy of the tumor lesion. The biopsy provided diagnostic confirmation of low-grade glioma in the anatomopathological study. Considering the topography of the lesion and the high potential for visual sequelae in the event of total surgical excision, it was decided that the patient would be followed up and administered chemotherapy with vincristine and carboplastine.
DISCUSSION
Neurofibromatosis was first published in 1768 as a case report of a patient with cutaneous fibromas supposedly inherited from his father4. Later, in 1882, Von Recklinghausen described the pathology in more detail, explaining in a more complex way the neural origin of the tumors. In 1940, Davis described optic nerve glioma associated with neurofibromatosis5.
This is a generic designation for a spectrum of three diseases of autosomal dominant genetic origin (AD)-neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and schwannomatosis. NF1, the most common form of the three, is caused by a change in a single gene located on chromosome 176, which leads to changes in the synthesis of neurofibromin-a protein involved in cell differentiation, control of survival, proliferation, and mitogenesis of the nerve tissues and integuments, skeletal tissues, and cardiovascular system7. This protein was detected by Nordlund et al. in all parts of the brain, especially in neurons with more extensive projections8.
It has a prevalence of approximately 1/3000 live births, with no preference for sex, and is inherited by one parent in approximately 50% of the cases. In other cases, it is not related to family history, which suggests a high incidence of new mutations1. The main clinical features of NF1 are café au lait macules, dermal and plexiform neurofibromas, axillary or inguinal effelids, and Lish nodules8.
Most symptomatic children present ophthalmic changes at diagnosis; the changes range from low visual acuity to strabismus, afferent pupillary defect, optic nerve atrophy, papilledema, and changes in color visualization. These symptoms are frequent in the groups aged below 7 or 8, and it is essential that children with NF1 in this age group have regular ophthalmologic examinations9.
Optic nerve gliomas are among the tumors most frequently found in NF110. Of these, astrocytoma is the most frequent11. The association of optic pathway glioma and NF1 is generally found in young children under 10 years of age12. In the case presented in this report, the tumor had extended beyond the left nucleocapsular region and cerebral peduncle, which explains the low vision perceived in the examination.
The diagnosis of NF1 is primarily clinical and is done using the National Institutes of Health diagnostic criteria for NF1, which are highly sensitive and specific for the vast majority of patients with the disease13 (Chart 1).

To date, there is no specific medication available to treat or prevent the characteristic lesions of neurofibromatosis. Treatment should be evaluated on a case-by-case basis, taking into account the location of the tumors, symptomatology, and their growth rate. Therapeutic options range from observation to chemo or radiotherapy or total or partial surgical resection.
NF1 is a disease that can present with a wide range of clinical ophthalmic symptoms, and the ophthalmologist is often the first professional to suspect its presence. Although there is no specific treatment or prevention of the disease, its early diagnosis is important for implementing an adequate treatment approach and consequent preservation of the vision of affected patients as well as for the guidance and genetic counseling of their relatives.
REFERENCES
1. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89(1):1-6.
2. Datson MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8(3):415-28.
3. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-8.
4. Arkenside M. Observation on cancer. Trans Med Soc Lond. 1768; 1:64-92.
5. Geller M, Bonalumi AF. Neurofibromatose. In: Carakushansky G. Doenças genéticas em pediatria. 1° ed. Rio de Janeiro: Guanabara Koogan; 2001.
6. Barker D, Wright E, Nguyen K, Cannon L, Fain P, Goldgar D, et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science. 1987; 236(4805):1100-2.
7. Suchmacher M. Neurofibromatose tipo 1: revisão sistemática da literatura - 1a parte. Atualidades Médicas. 2018;2(4):171-81.
8. Norduland M, Gu X, Shipley MT, Ratner N. Neurofibrin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci. 1993;13(4):1588-600.
9. Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol, 2006;13(1):2-7.
10. Stern J, DiGiacinto GV, Housepian EM. Neurofibromatosis and optic glioma: clinical and morphological correlations. Neurosurgery. 1979;4(6):524-8
11. Mukai K, Kitamura K, Asano N, Ohshima T, Hondo H, Matsumoto K. [Multifocal gliomas in cerebral hemisphere associated with von Recklinghausen’s disease: case report]. No Shinkei Geka. 1989;17(2):197-202.Japanese.
12. Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1 associated optic nerve glioma in mice. Ann Neurol. 2005; 57(1):119-27.
13. Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol 2013;115:939-55.
AUTHOR’S INFORMATION
Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
June 24, 2021.
Accepted on:
August 25, 2021.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket