

Victória d’Azevedo Silveira; Thaís Saorin Conte; Patrícia Ioschpe Gus
DOI: 10.17545/eOftalmo/2021.0029
RESUMO
Nas últimas décadas, com o advento de uma ampla gama de ferramentas diagnósticas sofisticadas e com o desenvolvimento de terapias gênicas para diversas condições antes intratáveis, interesse crescente vem sendo voltado às doenças oculares herdadas. No atual cenário, o oftalmologista geral deve ser capaz de reconhecer a presença ou associação de uma condição genética ou hereditária no caso de algum paciente e fornecer orientação quanto às possibilidades diagnósticas e terapêuticas vigentes, porém o aconselhamento genético ao paciente e/ou família deve ser fornecido por especialistas, como geneticistas e/ou oftalmologistas com título em aconselhamento genético, portanto realizar o encaminhamento para este fim.
Palavras-chave: Córnea; Genética; Oftalmologia.
ABSTRACT
In recent decades, the advent of a wide range of sophisticated diagnostic tools and the development of gene therapies for several previously intractable conditions has promoted the growing interest in inherited eye diseases. In the current scenario, general ophthalmologists must be able to recognize the presence or association of a genetic or hereditary condition in any patient’s case and provide guidance on current diagnostic and therapeutic possibilities, but genetic counseling for patients and their families must be provided by specialists, such as geneticists or ophthalmologists qualified in genetic counseling. Therefore, the patient should be referred to a specialist.
Keywords: Cornea; Genetics; Ophthalmology.
INTRODUÇÃO
O Hospital de Clínicas de Porto Alegre (HCPA) é referência em Doenças Metabólicas Herdadas para todo o Brasil, destacando-se em colaboração de experiências e atendimentos para toda a América Latina. Embora individualmente pouco prevalentes (na sua maioria, 1:100.000 nascimentos), o conjunto destas tantas doenças atinge 1:800 nascidos vivos. Esta prevalência se aproxima da síndrome de Down, a mais comum síndrome cromossômica, o que é altamente relevante para a saúde pública1-3.
O Serviço de Oftalmologia do HCPA apresenta importante interface com o Serviço de Genética Médica. Atendemos seus pacientes para auxiliar no diagnóstico incerto, para corroborar na aderência e efetividade dos tratamentos, e para diagnosticar complicações oculares. Da mesma forma, somos referência em transplante de córneas no Estado do Rio Grande do Sul, permitindo o atendimento das mais variadas patologias corneanas.
O presente artigo pretende colaborar com o conhecimento do leitor acerca de nossa experiência com as doenças oculares da córnea, sejam elas consequências das doenças genéticas sistêmicas, ou sejam elas primariamente oculares, mas de forte caráter geneticamente herdado. Priorizaremos as mais prevalentes.
Acometimento primariamente corneano:
1.1 Distrofias
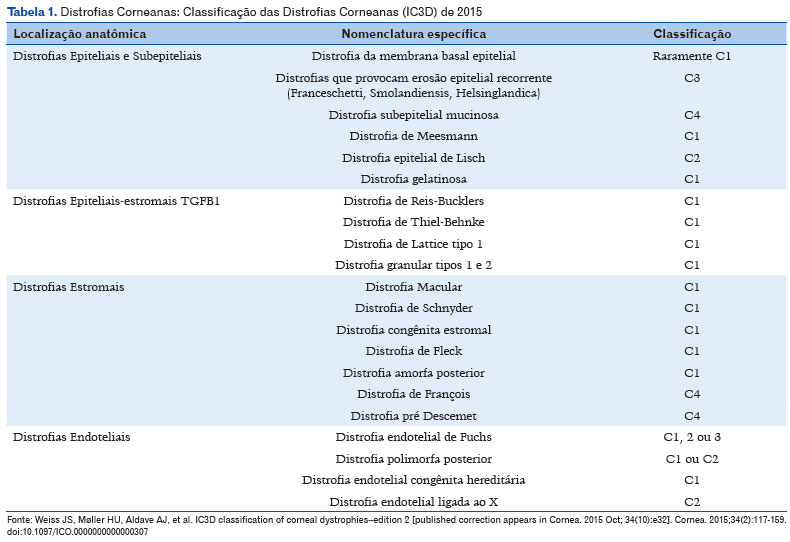
Caracteriza-se por um grupo de doenças bilaterais, simétricas, lentamente progressivas e cuja evolução independe de interferências ambientais. São tradicionalmente classificadas de acordo com a estrutura corneana primariamente acometida, embora mais de uma camada possa estar envolvida ao longo da evolução: epiteliais e subepiteliais, camada de Bowman, estromais, da Descemet e endoteliais1-3.
Análises genotípicas vêm alterando conceitos e introduzindo a evidência de heterogeneidade genética, fenotípica e de expressividade variável. Para exemplificar, algumas distrofias podem ser unilaterais, outras assimétricas, e outras ainda não dispõem comprovação laboratorial. Dentro deste novo paradigma, variações em um único gene podem acarretar diferentes fenótipos, como é o caso do gene TGFBI, e algumas distrofias podem ser causadas por variação em diferentes genes, como ocorre com a distrofia de Meesmann1-3.
Em 2008, a Sociedade Internacional de Córnea estabeleceu uma nova classificação para as distrofias corneanas, baseando-se em critérios anatômicos e reunindo-as em subgrupos de acordo com a base genética comum. Dividiu-as também em categorias:
• Categoria 1: gene mapeado e mutações específicas identificadas.
• Categoria 2: um ou mais loci cromossômicos mapeados, porém o gene necessita ser identificado.
• Categoria 3: ainda não foi identificado um loco cromossômico.
• Categoria 4: distrofia nova/suspeita ou já conhecida, porém com evidência fraca.
No ano de 2015, essa classificação sofreu algumas modificações em termos de classificação anatômica. As distrofias eram anteriormente classificadas de acordo com a lamela corneana mais afetada. No entanto, percebeu-se que o mais importante é saber qual a célula primária (de origem), uma vez que costumam afetar mais de uma lamela corneana.
A classificação atual das distrofias está resumida na Tabela 1. Não descreveremos uma a uma, tendo em vista a complexidade do assunto, o qual necessitaria de um capítulo a parte3.
1.2 Ectasias corneanas
O ceratocone é uma ectasia corneana assimétrica, progressiva e predominantemente bilateral, clinicamente caracterizada por afinamento corneano e astigmatismo assimétrico e com início na adolescência. É considerada multifatorial, tendo influência genética e ambiental, cuja prevalência tem aumentado com a possibilidade de diagnóstico precoce (pode variar de 1:50 em áreas de casamentos consanguíneos e clima quente e seco, como na Índia e no Irã, até 1:300 em áreas antes consideradas de baixo risco e prevalência, como a Holanda. Parece existir forte correlação entre surgimento de ceratocone e o hábito de coçar os olhos, como em pacientes alérgicos4-10.
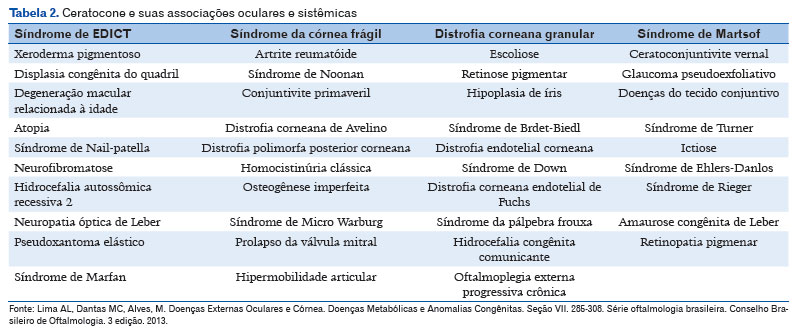
A história familial varia entre 5 e 20%, com média de 12 a 13%, em grandes estudos populacionais4-11. O ceratocone é considerado uma condição genética heterogênea, com penetrância incompleta e expressividade variável. A maioria dos casos familiais é de herança autossômica dominante com expressividade variável. Muitos genes estão envolvidos, incluindo: homeobox VSX1, SOD1, TGFB1, MIR 184, COL4A3/COL4A4 e FLG. Não é incomum a associação de ceratocone com outras doenças oculares e sistêmicas (Tabela 2)4-10.
Atualmente, as evidências de componente genético indicam uma atenção mais cautelosa aos familiares de pacientes acometidos pela doença. O estudo tomográfico e topográfico da córnea dos familiares é útil para identificar casos subclínicos e possibilitar diagnóstico precoce e tratamento efetivo da piora clínica4-10.
Ceratocone posterior
Caracteriza-se por indentação da córnea posterior em região central ou paracentral sem que ocorra a protrusão da superfície anterior da córnea, típica do ceratocone. É classificado como uma anomalia congênita, pois está associado à anteriorização da membrana de Descemet, presente desde o nascimento. Costuma ser estável e pode provocar astigmatismo irregular e ambliopia. A maioria dos casos é unilateral e esporádico11.
1.3 Megalocórnea
É caracterizada pelo diâmetro horizontal de 13 mm ou mais, bilateral, congênito e não progressivo. Cerca de 90% dos pacientes acometidos são do sexo masculino12,13.
Sugere-se que ocorra por alteração na produção de colágeno, uma vez que pode estar associada com síndrome de Marfan. Também pode se associar a goniodisgenesia, catarata, ectopia lentis, glaucoma congênito, dentre outras. É tipicamente uma condição autossômica recessiva ligada ao X por mutação no gene CHRDL1. Já foi relatada, entretanto, herança autossômica dominante12,13.
Faz-se diagnóstico diferencial com glaucoma congênito pela medida da pressão intraocular e pelos achados de biomicroscopia e do nervo óptico. Pode-se também utilizar ecografia para avaliar o comprimento axial, que nestes casos será normal12,13.
1.4 Microcórnea
Caracterizada por diâmetro corneano menor que 10mm ou menor que 9 mm em recém-nascidos. Pode ser unilateral ou bilateral. Ocorre por mutação no gene PAX6, cuja herança pode ser autossômica dominante ou recessiva. Aproximadamente 20% dos pacientes desenvolvem glaucoma ao longo da vida. Pode ocorrer de forma isolada ou associada a outras condições oculares, como catarata congênita e disgenesia do segmento anterior, ou sistêmicas, como síndrome alcóolica fetal, acondroplasia e síndrome de Ehler-Danlos12,13.
1.5 Córnea Plana
Caracteriza-se por raio de curvatura corneana menor do que 43 dioptrias. Um achado patognomônico é a curvatura da córnea igual à da esclera. Ocorre por mutação no gene KERA, cuja herança pode ser tanto autossômica dominante, quanto recessiva12,13.
Este gene codifica proteoglicanos importantes para a disposição das fibrilas de colágeno na córnea. Geralmente está associada a altos graus de hipermetropia (acima de 10 dioptrias) e à câmara rasa, predispondo a glaucoma de fechamento angular. Pode ser um achado isolado ou fazer parte da síndrome de Ehler-Danlos12,13.
1.6 Embriotoxon Posterior
Caracteriza-se por uma linha de Schwalbe (junção entre o trabeculado e o final da membrana de Descemet) espessada e anteriorizada, podendo ser observada a ectoscopia. Costuma ser um achado bilateral e de herança dominante, isolado ou associado a outras anomalias do segmento anterior, incluindo síndrome de Axenfeld-Rieger12,13.
1.7 Síndrome de Axenfeld-Rieger
Representa um espectro de condições causadas pelo desenvolvimento anômalo de estruturas do segmento anterior derivadas da crista neural. Pode cursar com microcórnea, embriotóxon posterior, hipoplasia iriana, corectopia, ectrópio uveal, glaucoma, dentre outras alterações. É bilateral, não tem predileção por sexo, apresenta herança autossômica dominante em 75% dos casos, podendo também ser esporádica. Pode ser causada por mutação nos genes PAX6, FOXC1 e PITX2. Associa-se ou não a alterações sistêmicas, incluindo hipoplasia maxilar, hipertelorismo e anormalidades dentárias, dentre outras12,13.
1.8 Anomalia de Peters
Caracterizada por ausência de membrana de Descemet e endotélio em uma porção da córnea, causando opacidade central ou paracentral desde o nascimento. Ocorre por defeito na migração de células da crista neural, predominantemente bilateral. A maioria dos casos ocorre de forma esporádica, embora possa ocorrer por herança autossômica dominante ou recessiva. Metade dos pacientes desenvolve glaucoma por anomalias angulares associadas. Divide-se em:
- Tipo I: ocorre adesão iridocorneana. A opacidade geralmente é avascular central ou total. Gerada por mutação dos genes PITX2, FOXC1, CYP1B1 e PAX6, é a de melhor prognóstico.
- Tipo II: ocorre adesão corneolenticular, cursando com catarata. A opacidade geralmente é vascularizada, podendo ser central ou total. Ocorre por mutação do gene FOXE3.
- Síndrome Peters plus: quando está associada a condições sistêmicas, incluindo baixa estatura, fenda palatina, alterações cardíacas, dentre outras12,13.
1.9 Esclerocórnea
Condição não progressiva e não inflamatória em que a córnea torna-se uma continuidade da esclera. Pode limitar-se à periferia ou acometer toda a extensão corneana. Costuma ser esporádica, mas pode ser herdade de forma recessiva ou dominante12,13.
1.10 Dermoide congênito
É um tumor sólido e benigno, unilateral ou bilateral, que apresenta pouco ou nenhum crescimento. É um coristoma, uma vez que contém elementos celulares que não estão usualmente presentes na superfície ocular, como folículos pilosos e glândulas sebáceas. Os casos unilaterais são esporádicos e predominantemente superficiais, enquanto os bilaterais estão associados à síndrome de Goldenhar, que se caracteriza pelo dermoide congênito, apêndice auricular e anormalidades vertebrais. Nesta, pode existir extensão do dermoide até o estroma profundo corneano, ou até acometimento de toda a extensão do segmento anterior12,13.
Secundárias a acometimento sistêmico:
Diversas condições sistêmicas podem cursar com acúmulo de substâncias na córnea, afetando a sua transparência. Quando primeiramente identificadas pelo oftalmologista, pode-se contribuir para o diagnóstico e salvar vidas12.
2.1 Mucopolissacaridoses
As mucopolissacaridoses (MPS) são um grupo de doenças lisossômicas causadas pela deficiência de enzimas que metabolizam os glicosaminoglicanos (GAGs), acarretando em seu acúmulo e prejuízo no metabolismo celular multissistêmico. Apresentam extensa variação fenotípica e são classificadas em 6 grupos. Na maioria dos casos, o padrão de herança é autossômico recessivo, com exceção da MPS tipo II, ligado ao X12,15,16.
A apresentação fenotípica inclui características faciais grosseiras, baixa estatura, organomegalia, displasia esquelética, doença cardíaca, pulmonar, oftalmológica, deficiência mental e alterações comportamentais em variáveis graus12,15,16.
É de extrema importância que o oftalmologista já tenha visto alguma imagem destes pacientes, pois só assim poderá pensar no diagnóstico. O fenótipo vai-se agravando com o tempo, e alguns pacientes falecem jovens. As alterações oculares são variadas, incluindo alta hipermetropia, opacidade corneana, espessamento escleral, glaucoma e pseudo-glaucoma, distrofia retiniana e atrofia óptica12,15,16.
Observa-se opacidade acinzentada e homogênea, em graus variáveis, à biomicroscopia. Perde-se a transparência de forma difusa, bilateral e lentamente progressiva, afetando a córnea uniformemente, de limbo a limbo, e tornando-a mais rígida, o que aumenta os valores da pressão intraocular e tende a hipermetropizar o paciente. Identifica-se aumento do volume dos ceratócitos e alteração das fibrilas estromais de colágeno à anatomopatologia12,15,16.
A turvação é causa importante de fotofobia e perda de acuidade visual corrigida, sendo evidenciada nos subgrupos de MPS IH (Hurler), MPS IS (Scheie), MPS IH / S (Hurler / Scheie), MPS IV (Morquio) e MPS VI (Maroteaux-Lamy). A turvação corneana é mais frequente na MPS IH e, inclusive, nos fenótipos atenuados da MPS I, sendo encontrada em 70% dos pacientes12,15,16.
Estudo cooperativo que avaliou transplantes realizados nestes pacientes confirma que a pressão intraocular retorna aos valores normais na maioria das córneas transplantadas, assim como a melhora da visão e a permanência da transparência do enxerto. São resultados impactantes na qualidade de vida. Entretanto, a terapia de reposição enzimática sistêmica pode não evitar a opacificação corneana16.
2.2 Doença de Fabry
A doença de Fabry é um defeito congênito do metabolismo dos glicosfingolipídeos, secundário à atividade deficiente da enzima α-galactosidase, e apresenta padrão de herança ligado ao X. Já foram descritas mais de 200 mutações do gene GLA, que codifica a α-galactosidase A12,17,19.
Os achados oculares detém papel importante na doença de Fabry, já que são uma das primeiras manifestações clínicas e costumas se manifestar na segunda década de vida. A córnea verticilata é o resultado de depósitos nas lâminas basais do epitélio. Outros achados oculares incluem: catarata, aneurisma conjuntival e edema de papila, dentre outros12,17,19.
O início e a progressão de manifestações são altamente variáveis, sendo que pacientes com formas mais agressivas costumam apresentar dor periférica intensa e frequentemente progridem para falência de múltiplos órgãos ainda na primeira década de vida. Além das alterações oculares, observa-se acroparestesias, angioqueratomas, hipoidrose, surdez, insuficiência renal e complicações isquêmicas cardíacas e cerebrais12,17,19.
Problemas comportamentais podem ser o primeiro sinal em adolescentes e adultos, algumas vezes diagnosticados como doentes psiquiátricos12,17,19.
Cistinose
Condição caracterizada pelo acúmulo de cristais de cistina especialmente nos olhos e nos rins por defeito no gene CTNS. É uma desordem metabólica rara e de herança autossômica recessiva. Pode ser dividida na forma infantil, grave e que causa insuficiência renal e morte na 1a década de vida, ou intermediária e juvenil. Os cristais promovem grande fotossensibilidade, mas não costumam baixar significativamente a acuidade visual. O tratamento das manifestações oculares é feito com colírio de cisteamina em múltiplas aplicações diárias, se possível próximas de dez12,19.
Doença de Wilson
Caracterizada por depósito hepático de cobre, em alguns casos, no cérebro e na membrana de Descemet, os quais são geralmente acometidos em conjunto. É uma condição autossômica recessiva que afeta o gene ATP7B. O clássico sinal é o anel de Kayser-Fleisher periférico da Descemet, marrom-dourado, em sua maioria. Pode ser necessário transplante de fígado para controle da doença. O achado corneano é útil para monitorar a resposta terapêutica, pois tende a sumir com o controle da doença12,19.
DISCUSSÃO
Os avanços na área da genética médica acontecem rapidamente nos tempos atuais. Surgem novos paradigmas e conceitos, permitido uma melhor compreensão das doenças, alterando prognósticos e permitindo melhor aconselhamento genético para inúmeros pacientes e suas famílias.
Dúvida comum repousa sobre a solicitação de testes genéticos e seu real benefício nas doenças primariamente corneanas. Os testes genéticos devem ser solicitados e interpretados por quem entende do assunto, pois os resultados são amplos e podem trazer informações inesperadas, bem como causar ansiedade. Dessa forma, quando indicado, sugere-se referir o paciente a um especialista em genética para avaliar a indicação e a necessidade deste. Deve-se sempre assegurar que o paciente procure um profissional, e não um laboratório diretamente, e que receba sempre uma cópia da sua testagem, quando realizada20.
Do ponto de vista oftalmológico, o diagnóstico e tratamento seguem baseados em critérios clínicos, diferentemente dos avanços recentes da terapia gênica para distrofias de retina causadas pelo gene RPE65. O futuro repousa sobre a possibilidade de terapia gênica com a introdução de sequências de DNA de efeito terapêutico de acordo com cada condição clínica, permitindo corrigir defeitos genéticos bastante específicos nas doenças monogênicas ou, pelo menos, minimizar danos em doenças poligênicas20.
O conhecimento da existência destas doenças pelo oftalmologista é de extrema relevância, uma vez que ele pode ser o primeiro médico ao qual os pacientes têm acesso. Muitas doenças genéticas podem ser tratadas, e os melhores resultados para a prevenção de complicações ocorrem quando o tratamento é precoce. Pode-se prevenir o surgimento ou a evolução dos defeitos oculares e sistêmicos, assim como proteger a vida dos pacientes.
Embora ainda não disponhamos de terapias gênicas dirigidas para a imensa maioria das condições genéticas oculares, a investigação diagnóstica é a melhor indicação. Em nossa instituição, casos suspeitos de síndromes que cursam com acometimen to sistêmico são encaminhados imediatamente para a Genética Médica, enquanto casos que cursam exclusivamente com acometimento ocular são diagnosticados e acompanhados pelo oftalmologista. Existem laboratórios especializados que realizam painéis diagnósticos abrangendo diversas patologias. Algumas das patologias para as quais podemos oferecer testagem genética em nossa instituição incluem: distrofia corneorretiniana cristalina de Bietti, espectro de microftalmia, anoftalmia e coloboma, síndrome de Alagille, mucopolissacaridose tipo IIIC, síndrome de Stickler, dentre inúmeras outras.
REFERÊNCIAS
1. Klintworth GK. Genetic disorders of the cornea: from research to practical diagnostic testing. Clin Exp Ophthalmol. 2005;33(3):231-2.
2. Vincent AL, Patel DV, McGhee CN. Inherited corneal disease: the evolving molecular, genetic and imaging revolution. Clin Exp Ophthalmol. 2005;33(3):303-16.
3. Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, et al. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015;34(2):117-59.
4. Godefrooij DA, de Wit GA, Uiterwaal CS, Imhof SM, Wisse RP. Age-specific Incidence and Prevalence of Keratoconus: A Nationwide Registration Study. Am J Ophthalmol. 2017 Mar;175:169-72.
5. Shetty R, Kaweri L, Pahuja N, Nagaraja H, Wadia K, Jayadev C, et al. Current review and a simplified “five-point management algorithm” for keratoconus. Indian J Ophthalmol. 2015;63(1):46-53.
6. Hashemi H, Heydarian S, Yekta A, Ostadimoghaddam H, Aghamirsalim M, Derakhshan A, et al. High prevalence and familial aggregation of keratoconus in an Iranian rural population: a population-based study. Ophthalmic Physiol Opt. 2018;38(4):447-55.
7. Hashemi H, Heydarian S, Hooshmand E, Saatchi M, Yekta A, Aghamirsalim M, et al. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea. 2020;39(2):263-70.
8. Godefrooij DA, de Wit GA, Uiterwaal CS, Imhof SM, Wisse RP. Age-specific Incidence and Prevalence of Keratoconus: A Nationwide Registration Study. Am J Ophthalmol. 2017 Mar;175:169-72.
9. Shetty R, Kaweri L, Pahuja N, Nagaraja H, Wadia K, Jayadev C, et al. Current review and a simplified “five-point management algorithm” for keratoconus. Indian J Ophthalmol. 2015;63(1):46-53.
10. Hashemi H, Heydarian S, Yekta A, Ostadimoghaddam H, Aghamirsalim M, Derakhshan A, et al. High prevalence and familial aggregation of keratoconus in an Iranian rural population: a population-based study. Ophthalmic Physiol Opt. 2018;38(4):447-55.
11. Gus PI, Araújo BS, Zelanis S, Schmalfuss TR, Marinho DR. Posterior keratoconus and iris atrophy: a fortuitous association? Arq Bras Oftalmol. 2019;82(1):68-71.
12. Lima AL, Dantas MC, Alves, M. Doenças Externas Oculares e Córnea. Doenças Metabólicas e Anomalias Congênitas. Seção VII. 285-308. Série oftalmologia brasileira. Conselho Brasileiro de Oftalmologia. 3 edição. 2013.
13. Dantas AM, Sallum JMF. Embriologia, genética e malformações do aparelho visual. Série oftalmologia brasileira. Conselho Brasileiro de Oftalmologia. 3 edição. 2013.
14. Del Longo A, Piozzi E, Schweizer F. Ocular features in mucopolysaccharidosis: diagnosis and treatment. Ital J Pediatr. 2018;44(Suppl 2);125.
15. Villas-Bôas FS, Fernandes Filho DJ, Acosta AX. Achados oculares em pacientes com mucopolissacaridoses. Arq Bras Oftalmol. 2011;74(6):430-4.
16. Ohden KL, Pitz S, Ashworth J, Magalhães A, Marinho DR, Lindahl P, et al. Outcomes of keratoplasty in the mucopolysaccharidoses: an international perspective. Br J Ophthalmol. 2017;101(7):909-12.
17. Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A. Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Exp Ophthalmol. 2005;33(2):164-8.
18. Cordeiro CA, Oréfice F, Lasmar EP, Santos HH, Valadares ER. Córnea verticilata - marcador clínico da doença de Fabry: relato de caso. Arq Bras Oftalmol. 2007;70(4):701-5.
19. Poll-The BT, Wenniger-Prick CJMB. The eye in metabolic diseases: clues to diagnosis. Eur J Paediatr Neurol. 2011;15(3):197-204.
20. Recommendations for Genetic Testing of Inherited Eye Diseases. American Academy of Ophthalmology. 2014. Available at: https://www.aao.org/clinical-statement/recommendations-genetic-testing-of-inherited-eye-d
INFORMAÇÃO DOS AUTORES
Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
21 de Julho de 2020.
Aceito em:
15 de Dezembro de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket