

Maria Isabel Lynch Gaete1; Ana Carolina Visco de Almeida2; Talita Raquel Sampaio Patriota3; Diogo Wagner Galindo Vaz Veras de Queiroz4; Jonielly Costa Vasconcelos de Santana5
DOI: 10.17545/eoftalmo/2018.0015
ABSTRACT
Mitochondrial myopathy or Kearns–Sayre syndrome is a rare disease with a typical triad: (1) onset at <20 years of age, (2) chronic progressive external ophthalmoplegia, and (3) pigmentary degeneration of the retina. Herein, we describe the case of a patient presenting with bilateral progressive ptosis, ocular hyperemia, and lacrimation for 6 years, which had been worsening for approximately 1 year, and who had a history of ptosis correction on two occasions at another institution, progressing toward exposure keratitis. This case highlights the need for caution when indicating ptosis correction to patients because the absence of Bell’s phenomenon due to mitochondrial myopathy and decreased muscle strength following this surgical treatment cause corneal exposure with insufficient lubrication and possible complications, such as exposure keratitis.
Keywords: Mitochondrial Myopathies; Kearns-Sayre Syndrome; Ophthalmoplegia, Chronic Progressive External.
RESUMO
A miopatia mitocondrial ou Síndrome de Kearns-Sayre é uma doença rara que contempla a tríade: indivíduos na segunda década de vida, oftalmoplegia externa progressiva crônica e degeneração pigmentária da retina. Neste relato, descreve-se o caso de um paciente que há 6 anos iniciou um quadro de ptose palpebral bilateral progressiva, hiperemia ocular e lacrimejamento com piora ao redor de 1 ano, com passado de correção de ptose em duas ocasiões em outro serviço, evoluindo com ceratite de exposição. O caso apresentado aponta a necessidade de cautela em relação à indicação cirúrgica da ptose palpebral, pois a ausência do Reflexo de Bell decorrente da Miopatia Mitocondrial e a diminuição da força muscular após correção cirúrgica da ptose propicia uma exposição corneana, com insuficiente lubrificação e complicações possíveis como ceratite de exposição.
Palavras-chave: Miopatias mitocondriais; Síndrome de Kearns-Sayre; Oftalmoplegia externa progressiva crônica.
RESUMEN
La miopatía mitocondrial o Síndrome de Kearns-Sayre es una enfermedad rara que contempla la siguiente tríada: individuos en la segunda década de vida, oftalmoplegia externa progresiva crónica y degeneración pigmentaria de la retina. En este relato, se describe el caso de un paciente que hace 6 años inició un cuadro de ptosis palpebral bilateral progresiva, hiperemia ocular y lagrimeo con empeoramiento alrededor de 1 año, con un pasado de corrección de ptosis en dos ocasiones en otro servicio, evolucionando para queratitis de exposición. El caso presentado señala la necesidad de cautela respecto a la indicación quirúrgica de la ptosis palpebral, dado que la ausencia del Reflejo de Bell consecuente de la Miopatía Mitocondrial y la disminución de la fuerza muscular tras corrección quirúrgica de la ptosis propician una exposición corneal, con insuficiente lubricación y posibles complicaciones tales como queratitis de exposición.
Palabras-clave: Miopatías Mitocondriales; Síndrome de Kearns-Sayre; Oftalmoplejía Externa Progresiva Crónica.
INTRODUCTION
Kearns–Sayre syndrome (KSS) was first described in 1958 by Kearns and Sayre, who reported two cases of young patients presenting with ocular changes associated with cardiac conduction disorders in the form of atrioventricular blocks, which can be fatal. In 1965, this condition was recognized as a syndrome and termed as KSS. In this rare condition, clinical manifestations, including ophthalmologic changes ranging from chronic progressive external ophthalmoplegia and retinal changes to exposure keratopathy, appear at approximately 20 years of age. Therefore, etiological investigation are required for early diagnosis and appropriate management of risk situations along with caution while recommending ptosis correction to patients with this syndrome.
METHODS
This is a descriptive case report. Data were obtained via anamnesis and additional examinations of the patient performed at Ophthalmological Service of Pernambuco (SEOPE, Brazil).
RESULTS
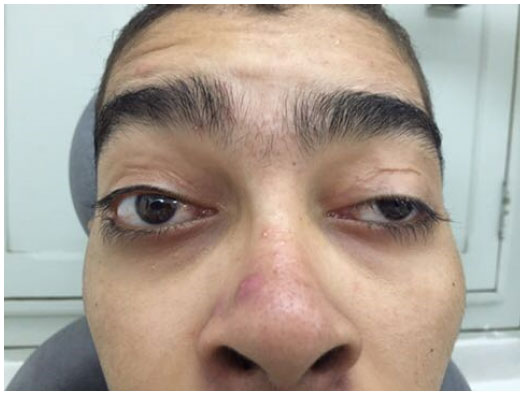
JCS, a 20-year-old male, visited SEOPE with a history of two corrections for bilateral ptosis at another institution 7 years prior to presentation. He reported progressive bilateral ptosis recurrence 6 years prior to presentation with ocular hyperemia and lacrimation, which had been worsening for 1 year. His examination revealed asymmetric ptosis with 7- and 5-mm palpebral fissures in the right (OD) and left (OS) eyes, respectively (Figure 1). Further, levator function was 2 mm for OD and 5 mm for OS, and the margin reflex distance was +1 for OD and 0 for OS. An examination of extraocular movements showed a notable limitation in the version and duction tests and an absence of Bell’s phenomenon. Further, best-corrected visual acuity (OU) was 0.5. Biomicroscopy revealed (1) OD showing relaxed conjunctiva, lower circular paracentral corneal opacity, and positive fluorescein staining and (2) OE showing lower linear paracentral corneal opacity and negative fluorescein staining. Tonometry showed a pressure of 10 mmHg OU. A funduscopic examination revealed optic discs without alterations, normal vessels, and equatorial and peripheral “salt and pepper”-type retinal pigmentation.
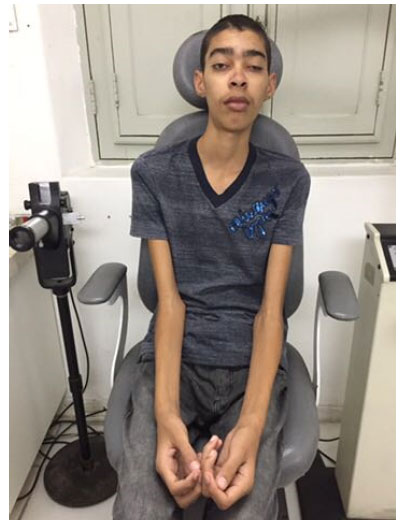
Additionally, a systemic clinical examination showed the following results: blood pressure, 110/80 mmHg; generalized muscle hypotrophy; and elongated appendicular limbs (Figure 2). Endocrinology and laboratory tests confirmed normal free T4 and TSH, elevated CPK (322 UI/L) and LDH (252 mg/dl), and normal aldolase levels. On the other hand, audiometry indicated bilateral sensorineural hearing loss that was moderate for the right and severe for the left ear. Although ECG revealed sinus rhythm with low incidence of supraventricular arrhythmia and right bundle branch block, head MRI demonstrated restricted diffusion in affected areas of the subcortical white matter in the occipital lobes and globus pallidus. These results suggest the presence of metabolic disease. The patient showed absence of oligoclonal bands when liquor electrophoresis was performed and a karyotype of 46 XY (male, no abnormalities). Further, muscle biopsy revealed granular muscle fibers. While modified Gomori staining proved the presence of ragged-red fibers, SDH staining revealed COX-negative and ragged-blue fibers. The muscle changes matched those in a mitochondrial myopathy pattern.
DISCUSSION
KSS is a rare disease of heterogeneous inheritance, with several types being observed to date. It is caused by mtDNA deletions, most of which are sporadic and believed to occur as germ-cell mutations or which occur very early during embryonic development. In addition, it is a multisystem disease that presents a triad of features: (1) onset at <20 years of age, (2) chronic progressive external ophthalmoplegia, and (3) pigmentary degeneration of the retina. Other symptoms include muscle weakness; chronic and progressive reduction of eye movements and ptosis; dysphagia; CNS dysfunction; ataxia; dementia, encephalopathy, or both; deafness; night blindness; syncope; and cardiac conduction defects1,2,3. Commonly, ptosis is the first clinical sign of KSS. Ptosis is usually bilateral and symmetrical; with its progression, the patient may adopt a posture of an elevated head and chin4,5. Decreased motility may go unnoticed until it becomes severe. In addition, there may be complaints of ocular hyperemia because of exposure keratopathy caused by Bell’s palsy6. In KSS, no findings are observed at birth; however, progressive external ophthalmoplegia and salt and pepper pigmentary retinopathy usually appears before the age of 20 years. The patient may present with cardiac conduction defects, often progressing toward sudden death7. Diagnosis is provided by muscle biopsy8, and treatment should be multidisciplinary with guarded prognosis. The disorder is usually progressive.
REFERENCES
1. Nonaka I. Mitochondrial disease. Curr Opin Neurol Neurosurg. 1992;5(5):622-32.
2. Souza CFM. Doenças da cadeia respiratória mitocondrial: um estudo clínico, bioquímico, histológico e molecular [Dissertação de mestrado]. Porto Alegre: Universidade Federal do Rio Grande do Sul (UFRGS); 2001. 100 p.
3. Schmitz K, Lins H, Behrens-Baumann W. Bilateral spontaneous corneal perforation associated with complete external ophthalmoplegia in mitochondrial myopathy (Kearns-Sayre syndrome). Cornea. 2003;22(3):267-70.
4. Cohen JM, Waiss B. Combination ptosis crutch and moisture chamber for management of progressive external ophthalmoplegia. J Am Optom Assoc. 1997;68(10):663-7.
5. Dias-Tosta E. Chronic progressive external ophthalmoplegia: I. Aquantitative histochemical study of skeletal muscles. Arq Neuropsiquiatr. 1988;46(2):133-42.
6. Fraunfelder FT, Roy FH, Randall J. Chronic progressive external ophthalmoplegia. In: Fraunfelder FT, Roy FH. Current Ocular Therapy. 5th ed. Philadelphia: W.B. Saunders; 2000. p. 208-10.
7. Cruz MW, André C, Hahn MD, Mattos JP, Maranhão Filho PA, Silva ES, et al. Síndrome de Kearns-Sayre: relato de caso com instalação rápida e documentação anátomo-patológica. Rev Bras Neurol. 1989;25(2):55-60.
8. Consoni Filho E, Oliveira ASB, Schmidt B. Musculatura extra-ocular: um músculo esquelético diferenciado. Arq Bras Oftalmol. 1994;57(6):394-9.





Funding: No specific financial support was available for this study.
CEP Approval: Not applicable
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
April 10, 2018.
Accepted on:
June 7, 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket