

Francisco Victor Carvalho Barroso1; Ana Laura Eloia Limão2; Rian Vilar Lima3; Samuel Montenegro4; Manoela Gondim5; João Crispim Moraes Lima Ribeiro2
DOI: 10.17545/eOftalmo/2024.0014
RESUMO
O objetivo desta revisão de literatura é estudar as alterações oculares em pacientes com Síndrome de Dandy-Walker. Uma revisão da literatura foi feita usando as bases de dados Embase, Medline, Lilacs e Scielo, pesquisando artigos usando as seguintes palavras gerais "Eye" e "Dandy-Walker" como termos de busca, sem limitar onde esses termos foram encontrados em diferentes seções do artigo. A busca encontrou 109 artigos e após critérios de exclusão 1.110 artigos. Foram selecionados 54 registros para leitura na íntegra e finalmente incluídos 22 estudos para descrever as associações. 57% de todos os pacientes da nossa amostra apresentaram alteração do segmento anterior e 42% alteração do segmento posterior. Estudos de alto impacto são necessários para melhor descrever as associações mencionadas.
Palavras-chave: Síndrome de Dandy-Walker; Mega cisterna magna; Anomalias oculares; Oftalmológica; Oftalmologia.
ABSTRACT
Through this literature review, we aimed to determine the ocular alterations in patients with Dandy-Walker syndrome. Embase, Medline, Lilacs, and Scielo databases were searched using the following key words "Eye" and "Dandy-Walker" without any limits. The search yielded 1110 articles, which was reduced to 109 articles after non-relevant articles were excluded. The full text of 54 articles were read, and 22 studies were finally evaluated. Approximately 57% and 42% of all the patients in our sample exhibited anterior segment and posterior segment alterations, respectively. However, high impact studies are required to better describe the associations between ocular manifestations and Dandy-Walker syndrome.
Keywords: Dandy-Walker syndrome; Mega cisterna magna; Eye abnormalities; Ophthalmologic; Ophthalmology.
INTRODUÇÃO
A síndrome de Dandy-Walker (SDW) é uma malformação rara da fossa posterior, presente em apenas 1 em 25.000 a 30.000 nascidos vivos1,2. As malformações da fossa posterior mais frequentes foram mega cisterna magna (31,8%) e malformação de Dandy-Walker (27%)3. A DWS foi descrita pela primeira vez em 1954. A SDW normalmente ocorre esporadicamente. Mantendo o desenvolvimento do vermis cerebelar como padrão, o DWS pode ser dividido em: 1. Malformação de Dandy-Walker; 2. Variante Dandy- Walker; e 3. Alargamento simples da cisterna da fossa posterior. Em vez disso, devem ser mencionadas anomalias físicas individuais. Muitos pacientes permanecem clinicamente assintomáticos durante anos, enquanto outros podem apresentar uma variedade de comorbidades que levam ao diagnóstico precoce4. A SDW tem sido associada a deleções do cromossomo 3 que causam dilatação cística no 4º ventrículo no 4º mês de vida fetal5. Assim, pode associar algumas características oculares como hipertelorismo, epicanto, glaucoma, anoftalmia, uma vez que também estão relacionadas a mutações no cromossomo 36. Outras mutações genéticas como ZIC, ZIC4, FOXC1, FGF17, LAMC1 e NID17-13 também foram relatados. Diferentes tipos de deleções dos cromossomos 3, 6 e 13 foram relatados. O cromossomo 13 possui intervalo de deleção principalmente na região 13q32.2-33.2, na qual os genes ZIC2 e ZIC513. G9a, também conhecido como Eucromatina histona metiltransferase 2 EHMT2, uma proteína lisina metiltransferase que introduz modificação de metilação em uma variedade de proteínas, incluindo histonas, também foi associada ao SDW. Ainda assim, nenhum deles está relacionado à embriogênese ocular13.
As características anatômicas clássicas são hipoplasia do vermis cerebelar, aumento anteroposterior da fossa posterior e dilatação cística do quarto ventrículo. A variante SDW foi definida devido à ausência de luxação do tentório, inversão torcular-lambdoidal ou rotação vermiana. O tratamento geralmente se concentra no alívio dos sintomas da hidrocefalia e da fossa posterior, muitas vezes incluindo intervenções cirúrgicas como derivação ventriculoperitoneal e cisto peritoneal. No entanto, falta literatura sistematizada sobre as manifestações oculares da SDW. Portanto, nosso objetivo foi revisar sistematicamente os casos relatados na literatura.
MÉTODOS
Revisão de literatura
Este estudo seguiu uma abordagem exploratório- descritiva e qualitativa utilizando os métodos de revisão sistemática do protocolo Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) 2020 (Figura 1) MEDLINE (via PubMed), Lilacs e Scielo (via BVS), Bancos de dados Embase e Web of Science. A busca dos artigos foi realizada utilizando os descritores apropriados para cada plataforma, sem considerar idioma ou recorte temporal específico. Os descritores (MeSH) "Síndrome de Dandy-Walker", "DWS", "mega cisterna magna", "eye", "eye anomalias", "oftalmológico", "oftalmologia", "oftalmológico", foram utilizados e cruzados utilizando o booleano operador E e OU. As referências de todos os estudos incluídos também foram revisadas para publicações adicionais.
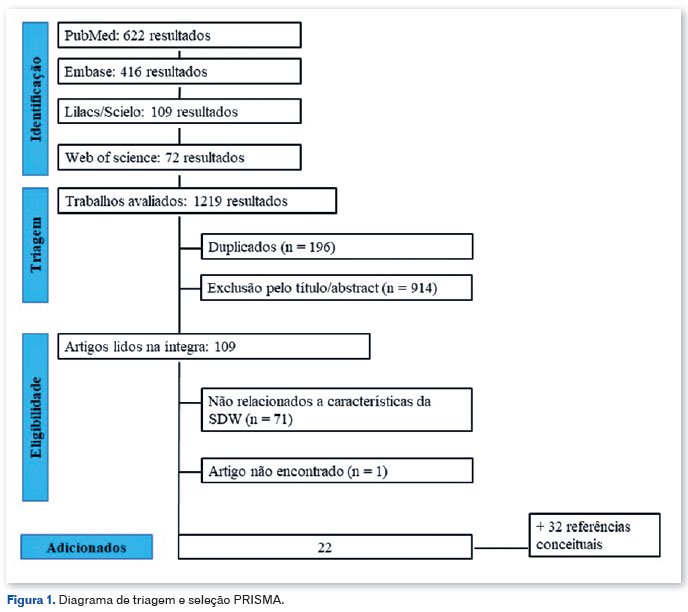
Critério de eleição
Inicialmente, foram identificados e submetidos para triagem 1.110 artigos, com análise independente por quatro pesquisadores e possíveis divergências sendo resolvidas pela avaliação de um quinto investigador. Após a exclusão das duplicatas, restaram 1.023 estudos, e seus títulos e resumos foram julgados com base no modelo PECOT para critérios de inclusão e exclusão da seguinte forma: (a) participantes: pacientes com DWS; (b) exposição: desenvolvimento de anomalias oculares; (c) controle: nenhum; (d) desfecho: tipo de anormalidade, segmento da anormalidade, genes associados; e (e) desenho do estudo: estudos observacionais. Os critérios de exclusão de acordo com as normas: (a) estudos que não relataram alterações oftalmológicas ou qualquer avaliação oftalmológica; (b) sobreposição de populações entre publicações; e (c) os textos completos ou os dados completos não estavam disponíveis após contato com os autores originais. Para garantir que a revisão incluísse todos os achados relevantes, estudos que relataram pacientes com anomalia de Dandy-Walker foram incluídos mesmo quando não confirmavam a evidência radiológica diagnóstica. A qualidade dos estudos não foi critério de exclusão, mas foi devidamente reconhecida nos resultados.
Extração de dados
Dois autores extraíram independentemente os dados de todos os estudos elegíveis. Para garantir a integridade e precisão das informações, as divergências foram discutidas e resolvidas por consenso com um terceiro revisor. Extraímos os seguintes dados dos estudos elegíveis: sobrenome do primeiro autor, ano e país de publicação, fontes de dados, tempo de acompanhamento, faixa etária, número de pacientes, idade média ou mediana, anomalias genéticas e anomalias oculares no segmento anterior e segmento posterior.
Análise de dados
As variáveis foram expressas qualitativamente. Foi adotada uma abordagem puramente descritiva (isto é, dados expressos como médias não ponderadas sempre que possível) para variáveis contínuas. Nenhuma análise estatística adicional foi realizada.
Aspectos éticos
Como os artigos analisados neste estudo estão disponíveis publicamente nas respectivas bases de dados e portais científicos, foi dispensada a apreciação ética.
RESULTADOS
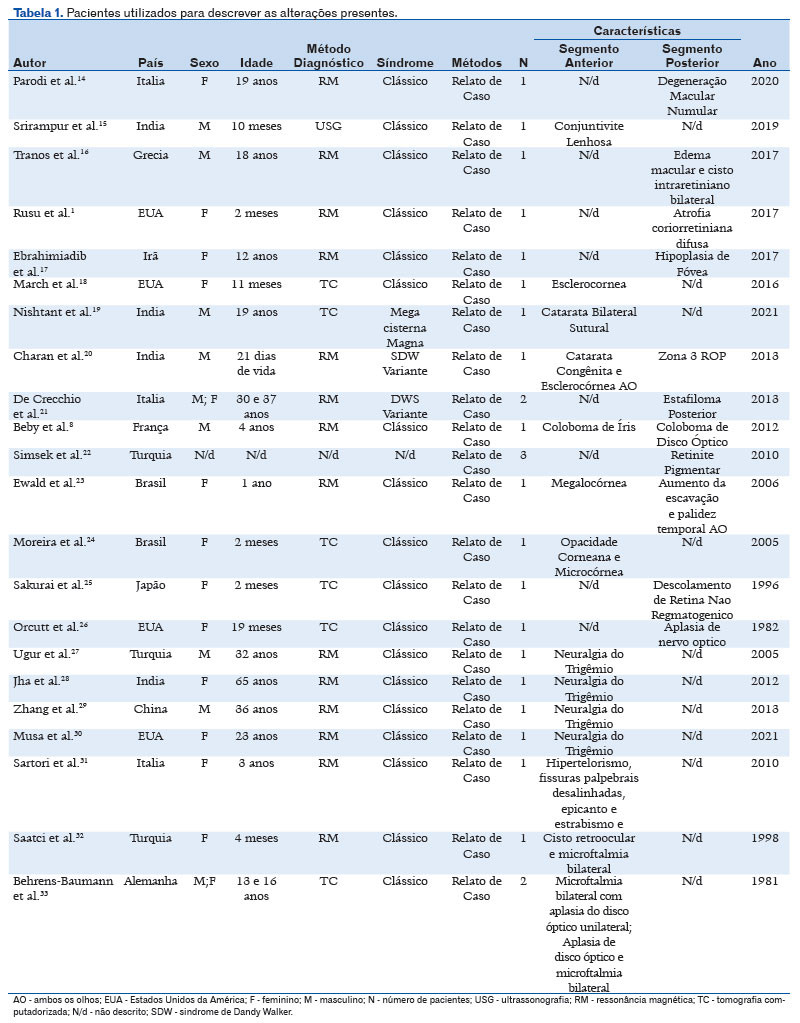
Foram selecionados 54 registros para leitura na íntegra e ao final foram incluídos 22 estudos. Um estudo foi uma série de casos de 2 pacientes e todos os demais foram relatos de casos, totalizando 26 casos analisados. Sessenta e seis porcento eram do sexo feminino, 34% do sexo masculino, 15,6 anos (DP ± 16,9 anos). Cinquenta e sete porcento da nossa amostra apresentou alteração do segmento anterior e 42% alteração do segmento posterior (Tabela 1). Algumas patologias encontradas foram descritas de acordo com a sequência da estrutura do segmento anterior e posterior. Os fenótipos diagnosticados encontrados foram microftalmia, hipotricose de cílios e sobrancelhas, hipertelorismo, sobrancelhas grossas e cílios longos, micro e megalocórnea, esclerocórnea, catarata bilateral, conjuntivite lenhosa e congênita, coloboma de íris e disco óptico, descolamento não regmatogênico, despigmentação macular numular, edema macular, miopia patológica e neuralgia do trigêmeo.
DISCUSSÃO
Na pesquisa foram relatadas algumas síndromes relacionadas à DWS e as seguintes alterações de órbita, anexos, alterações anteriores, posteriores e características neurooftalmológicas.
Associações sindrômicas
Alguns artigos descreveram a associação entre JS e DWS34-36. JS é uma doença autossômica recessiva, caracterizada por hipoplasia vermiana e anomalias do tronco cerebral, na imagem. Algumas características sistêmicas como hipotonia, atraso no desenvolvimento, ataxia, padrões respiratórios irregulares e movimentos oculares anormais também estão relacionados. Combinações de características adicionais, como polidactilia, coloboma ocular, distrofia retiniana, doença renal, fibrose hepática, encefalocele e outras malformações cerebrais definem subtipos clínicos. Sartori et al.37 relataram uma DWS associada a recém-nascidos com JS com hipertelorismo, fissuras palpebrais oblíquas, pregas epicânticas bilaterais, estrabismo e cílios longos. Além disso, foi demonstrado que a DWS está associada à síndrome de Down, síndrome PHACE (malformações cerebrais da fossa posterior, hemangiomas, anomalias arteriais, coarctação da aorta e defeitos cardíacos e anomalias oculares).
Síndrome; Síndrome de Ritscher-Schinzel, Síndrome de Walker Walburg, trissomia 18, síndrome de Joubert (SJ), síndrome de Wisconsin, Síndrome de Delleman38-41.
Anormalidades de órbita
Globo ocular
A microftalmia é uma das anomalias congênitas oculares mais comuns. Refere-se a um globo ocular pequeno e desorganizado devido ao defeito quase completo da vesícula óptica primária, o que leva a um olho ausente ou muito pequeno dentro da órbita.
O objetivo do tratamento é evitar a diminuição da fossa orbitária por expansão precoce.
A microftalmia e o cisto retrobulbar foram relatados por Saatci et al.32. Menina de 4 meses com cefalocele parietal atrésica e malformações oculares. Supunha-se que essas malformações eram devidas à falha no fechamento do saco óptico durante o desenvolvimento embriológico.
Anormalidades de anexos
Cílios e sobrancelhas
Boudghene-Stambouli et al.42 descreveram a síndrome KID (ceratite-ictiose-surdez) em uma menina nascida de pais não consanguíneos sem histórico familiar semelhante com hipotricose de cílios e sobrancelhas. Além disso, Pascual-Castroviejo et al.43 mostraram dois irmãos com síndrome oculocerebrocutânea (Delleman). Esta síndrome é caracterizada por anomalias congênitas que envolvem pele, órbita e sistema nervoso central (SNC). Um irmão apresentava um apêndice cutâneo ao redor da sobrancelha e hipoplasia do globo ocular. O segundo irmão apresentou cistos orbitais e apêndices palpebrais. Moreira et al.24 relatou hipertelorismo, sobrancelhas grossas e cílios longos.
Anormalidades do segmento anterior
Córnea
A megalocórnea é conhecida como um defeito congênito bilateral raro e não progressivo, caracterizado por aumento do diâmetro da córnea superior a 12,5mm a 13mm ao nascimento. Sua complicação como fibras zonulares fracas pode complicar a cirurgia de catarata que necessita de lentes intraoculares de 3 peças23. Como o defeito é anatômico, não há cura ou tratamento para o aumento da córnea. A intervenção mais comum é a cirurgia de catarata, com abordagem específica às necessidades individuais, ou a cirurgia para colocação de lente intraocular em forma de garra de íris44. Ewald et al.23 relataram uma menina de 1 ano e 9 meses de idade com DWS e megalocórnea no olho direito de 13mm e no olho esquerdo de 13,5mm. O exame de fundo de olho mostrou relação escavação/disco de 0,7 em ambos os olhos, palidez do disco óptico temporalmente em ambos os olhos e pressão intraocular de 16mmHg no olho direito e 14mmHg no olho esquerdo. O diâmetro anteroposterior foi de 20,55mm em ambos os olhos. Mas não foram encontradas estrias de Haab ou fotofobia, excluindo o diagnóstico de glaucoma congênito. A microcórnea é definida por uma anomalia congênita da córnea na qual o diâmetro horizontal da córnea é menor que o normal. É importante lembrar que nestes casos o globo tem dimensões normais. Foi proposto que o crescimento excessivo anormal das pontas anteriores do copo óptico durante o desenvolvimento do olho causa isso45. Moreira et al. relatado em uma menina de 2 meses e Charan et al.20,24 relataram um caso de microcórnea em um recém-nascido de 21 dias também com anomalias da córnea, como opacidade e microcórnea, e a tomografia computadorizada da cabeça sugeriu DWS. Um resultado semelhante relacionado à opacidade da córnea foi relatado por Stambolliu et al.46 já que 20% dos pacientes com catarata estavam associados à DWS durante a revisão da literatura. Além disso, March et al.18 relataram esclerocórnea em um recém-nascido de 21 dias.
Anormalidades do cristalino
Cataratas bilaterais foram relatadas por Charan et al.20 também e Nishtant et al.19 relataram um homem de 19 anos com catarata bilateral e defeitos de campo visual. O tipo de catarata foi classificado como sutural e a lesão do lobo occipital foi explicada como causa da lesão do campo visual.
A catarata sutural bilateral, por exemplo, é uma patologia congênita de origem genética que afeta as suturas em forma de Y do cristalino. A doença não costuma progredir e, geralmente, não interfere na acuidade visual do paciente. Geralmente, o tratamento não é necessário. Em alguns casos é cirúrgico47.
Anomalias conjuntivais
Conjuntivite neonatal foi relatada por Moreira et al.24 relatado em uma conjuntivite bilateral neonatal de 21 dias. A conjuntivite lenhosa é definida por uma forma rara de conjuntivite crônica e recorrente devido à deficiência da atividade do plasminogênio, promovendo pseudomembranas fibrinosas na conjuntiva tarsal e bulbar. Seu tratamento baseia-se no uso de fibrinolítico tópico seguido de excisão cirúrgica.
Srirampur et al.15 por exemplo, relatou conjuntivite lenhosa em uma criança de 10 meses de idade diagnosticada com DWS por ultrassonografia pré-natal na 34ª semana. A criança apresentou conjuntivite pseudomembranosa lenhosa principalmente no olho direito, sem outras manifestações oculares e o exame hematológico mostrou deficiência de plasminogênio.
O manejo terapêutico é longo, difícil e mal codificado. Este tratamento baseia-se na combinação de colírios com ciclosporina, heparina e corticosteroides. Aplicações locais e subconjuntivais de plasma fresco congelado podem estar associadas à melhoria da resposta terapêutica31.
Anormalidades da íris
Os colobomas congênitos da íris raramente precisam de reparo até o momento da cirurgia de catarata. Os colobomas ópticos resultam da falha no fechamento da fissura óptica durante a embriogênese. Eles podem envolver a íris e estruturas subsequentes ao nervo óptico. Coloboma de íris foi relatado por Beby et al.8 em um homem de 4 dias de idade com embriotoxon posterior bilateral. Os colobomas congênitos da íris variam consideravelmente em forma, tamanho e localização exatos de paciente para paciente, portanto, a execução do reparo cirúrgico precisa ser personalizada de acordo com o olho de cada paciente48.
Segmento posterior
Retina
Além disso, foi relatado que características segmentares posteriores estão associadas à síndrome de Dandy-Walker. Alterações nos achados retinianos, como descolamento não regmatogênico relatado por Sakurai et al.25 despigmentação macular numular14, e edema macular relacionado por Tranos et al.16. O descolamento não regmatogênico é a forma mais comum de descolamento de retina, ocorre quando o fluido vítreo entra através de uma ruptura ou buraco na retina, levando à separação da retina da coroide. Alguns descolamentos de retina podem ser tratados em ambiente clínico, aplicando laser na retina ou injetando uma bolha de gás no olho para recolocar a retina. Descolamentos de retina complexos, caracterizados por múltiplas rupturas da retina ou pela presença de tecido cicatricial da retina, são normalmente reparados usando vitrectomia via pars plana ou procedimento de flambagem escleral em uma sala de cirurgia49.
Nervo óptico
O coloboma do disco óptico pode ocorrer esporadicamente ou ser herdado com herança autossômica dominante. É o resultado da fusão anormal dos dois lados da extremidade proximal do copo óptico, compreendendo uma escavação em forma de tigela claramente demarcada do disco óptico, que é tipicamente descentralizada e mais profunda inferiormente50. Beby et al. 8 relataram um coloboma de disco óptico em um homem de 4 dias de idade. Ewald et al.23 relataram uma menina de 1 ano e 9 meses de idade com DWS com relação escavação/disco de 0,7 em ambos os olhos e palidez do disco óptico temporalmente em ambos os olhos. Behrens-Baumannet al.33 relataram dois irmãos com malformações oculares graves. A menina de 16 anos tem microftalmia bilateral com aplasia do nervo óptico direito e cisto de Dandy-Walker no cerebelo. O olho direito não possui disco óptico ou vasos retinianos. Seu irmão de 13 anos tem aplasia unilateral do nervo óptico com criptoftalmia e microftalmia contralateral. Aplasia do nervo óptico foi relacionada por Orcutt et al.26.
Mácula
Tranos et al.16 apresentou um homem de 18 anos com DWS encaminhado após diminuição da acuidade visual (20/30) nos últimos 3 meses. A fundoscopia revelou reflexo foveal anormal em ambos os olhos e a imagem com tomografia de coerência óptica de domínio espectral mostrou pequenos cistos intrarretinianos bilaterais com alguma melhora de função após dois meses com injeção intravítrea de triancinolona (0,05 cc de 40mg/ml). No entanto, quatro meses depois, o cisto intrarretiniano reapareceu.
Os cistos retinais podem ser únicos ou múltiplos, variando de diâmetro de dois a dez discos, e ocorrem em olhos com descolamento de retina de longa data. Raramente causam sintomas oculares ou visão central prejudicada. A maioria dos cistos se resolve espontaneamente com a reinserção da retina e melhor nutrição da retina. O tratamento cirúrgico da condição subjacente geralmente leva à resolução completa51.
Em relação a outras anomalias da retina, a retinite pigmentosa (RP) também foi descrita em nossa pesquisa. É uma retinopatia geneticamente heterogênea causada pela morte de células fotorreceptoras e atrofia do epitélio pigmentar da retina que eventualmente resulta em cegueira nos olhos bilaterais. À medida que a doença progride, o tratamento mais eficaz para as diferentes fases depende do número de células fotorreceptoras restantes. As modalidades de tratamento são: agente neuroprotetor, terapia genética, terapia com células-tronco, optogenética, retina artificial e fotobruxas químicas52.
RP foi descrito por Simsek et al.22 que relataram três irmãos com síndrome de Dandy-Walker associada à síndrome de Usher de pais turcos consanguíneos. Eles apresentavam retinite pigmentosa e perda auditiva, embora a síndrome de Usher seja caracterizada por esses sintomas, além de problemas ocasionais de equilíbrio.
Outra característica, uma hipoplasia foveal, que é definida por uma distorção da retina e que tem a perda completa da morfologia da fóvea. Ebrahimiadib et al.17 descreveram uma menina de 12 anos com história de SDW associada à perda crônica não progressiva da visão. Não havia história de erro refrativo e nenhum relato de ptose ou estrabismo na infância. A acuidade visual era 20/50 em ambos os olhos e o exame de fundo de olho revelou falta de reflexo foveal e leve tortuosidade dos vasos em ambos os olhos. Além disso, havia continuidade anormal das camadas internas da retina e ausência de fosseta foveal grau 2 vista bilateralmente pela tomografia de coerência óptica.
O estafiloma posterior (EP) é uma bolsa externa de uma região circunscrita do fundo posterior e tem sido considerada uma marca registrada da miopia patológica. Caracterizado por um afinamento escleral relativamente abrupto53. EP foi relatado por De Crecchio et al.21 aquele relacionado a dois irmãos diagnosticados com a variante SDW com olhos míopes altos. O primeiro foi um homem de 37 anos com história pregressa de doença de Hodgkin e trabeculectomia por pressão intraocular elevada 7 anos antes do diagnóstico de estafiloma posterior. Foi apresentado afinamento do epitélio pigmentar da retina na região da mácula em ambos os olhos e membranas vítreas na retina periférica. Além disso, a segunda era uma mulher de 30 anos com alta miopia em ambos os olhos com estafiloma posterior e membrana vítrea na periferia inferior e temporal. A degeneração da despigmentação macular numular também foi associada à SDW, conforme relatado por Parodi et al.14.
A despigmentação macular numular é definida por um distúrbio adquirido da pigmentação da pele, que é geralmente subdiagnosticado. É caracterizada por lesões numulares hipopigmentadas que aparecem no tronco em pessoas jovens. Várias opções de tratamento estão disponíveis, embora a clindamicina tópica e o peróxido de benzoíla tenham sido tradicionalmente usados. Bons resultados foram alcançados recentemente usando fototerapia ultravioleta B de banda estreita (NBUVB)54. Ademais, Rusu et al.1 relataram não perfusão vascular retiniana na retina temporal de ambos os olhos em irmãos de 2 meses de idade após a realização de angiografia com fluoresceína. O exame de fundo de olho revelou nervos planos e rosados, atrofia coriorretiniana difusa e manchas pigmentares na mácula de ambos os olhos.
Anormalidades neurolftalmo
Divisão oftálmica do nervo trigêmio
A neuralgia do trigêmeo (NT) é um distúrbio de dor neuropática raro, caracterizado por episódios recorrentes e paroxísticos de dor intensa e de curta duração, semelhante a um choque elétrico, ao longo da distribuição sensorial do nervo trigêmeo27. TN foi relatada por Ugur et al., Jha et al. e Zhang et al.27-29. Além disso, Musa et al.30 em uma mulher de 23 anos explicou que o acúmulo de líquido cefalorraquidiano no cisto da fossa posterior foi responsável pela compressão da raiz do nervo trigêmeo, na zona de entrada da raiz do trigêmeo, alguns milímetros após a entrada no nível da ponte. O manejo médico consiste em modalidades farmacológicas, com medicamentos antiepilépticos e intervenções reversíveis, como toxina botulínica tipo A e injeções de anestesia local. As terapias cirúrgicas consistem em diversos procedimentos periféricos e intracranianos27.
SDW e suas características oculares associadas não foram amplamente estudadas. Os casos mencionados ajudam a comunidade científica a esclarecer e explicar algumas alterações oculares na SDW. Mas são necessários estudos de alto impacto para melhor descrever essas características oculares.
REFERÊNCIAS
1. Rusu I, Gupta MP, Patel SN, Oltra E, Chan RV. Retinal vascular nonperfusion in siblings with Dandy-Walker variant. J AAPOS. 2016;20(2):174-7.
2. Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125-48.
3. Pira-Paredes SM, Montoya-Villada JH, Franco-Restrepo JL, Moncada-Velez M, Cornejo JW. [A phenotypic description of 26 patients with Ritscher-Schinzel syndrome (cranio-cerebello-cardiac dysplasia or 3C syndrome)]. Rev Neurol. 2017;64(11):481-488. Spanish.
4. Zamora EA, Das JM, Ahmad T. Dandy-Walker Malformation. [Updated 2023 Nov 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538197/
5. Delahaye A, Khung-Savatovsky S, Aboura A, Guimiot F, Drunat S, Alessandri JL, et al. 9. Am J Med Genet A. 2012;158A(10):2430-8.
6. Chi L, Zhong L, Lee D, Yu X, Caballero A, Nieman B, et al. G9a inactivation in progenitor cells with Isl1-Cre with reduced recombinase activity models aspects of Dandy-Walker complex. Biol Open. 2023;12(8):bio059894.
7. Beaman MM, Guidugli L, Hammer M, Barrows C, Gregor A, Lee S, et al. Novel association of Dandy-Walker malformation with CAPN15 variants expands the phenotype of oculogastrointestinal neurodevelopmental syndrome. Am J Med Genet A. 2023;191(11): 2757-2767.
8. Beby F, Des Portes V, Till M, Mottolese C, Denis P. Chromosome 6p25 deletion syndrome: report of a case with optic disc coloboma and review of published ophthalmic findings. Ophthalmic Genet. 2012;33(4):240-8.
9. Kotani T, Tsuda H, Ito Y, Nakamura N, Ushida T, Imai K, et al. Prenatal diagnosis of distal 13q deletion syndrome in a fetus with esophageal atresia: a case report and review of the literature. J Med Case Rep. 2022;16(1):481.
10. Santos BZ, Zancanela LB, Argemi CT, Chiquetti EMS. Terminal deletion of the short arm of chromosome 6 (6P25 3P 243): a literature review and case report of a Brazilian child/ Deleção terminal do braço curto do cromossomo 6 (6p25 3p 243): uma revisão de literatura e relato de caso de uma criança brasileira. Medicina (Ribeirao Preto, Online);55(2)abr 2022 ilus, tab. 2022.
11. Alp MY, Çebi AH, Seyhan S, Cansu A, Aydin H, Ikbal M. 22.5 MB DELETION OF 13q31.1-q34 ASSOCIATED WITH HPE, DWM, AND HSCR: A CASE REPORT AND REDEFINING THE SMALLEST DELETED REGIONS. Genet Couns. 2016;27(1):43-9.
12. Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037-42.
13. Ballarati L, Rossi E, Bonati MT, Gimelli S, Maraschio P, Finelli P, et al. 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet. 2007;44(1):e60.
14. Parodi MB, Arrigo A, Manitto MP, Bandello F. Nummular Macular Depigmentation in Dandy-Walker Syndrome. Retina. 2020;40(9): e46-e47.
15. Srirampur A, Ramappa M, Chaurasia S, Vemuganti G. Ligneous conjunctivitis in a Dandy-Walker syndrome: A rare case report. Indian J Ophthalmol. 2019;67(1):143-145.
16. Tranos P, Dervenis N, Kiouras S. Bilateral Macular Edema: A New Ocular Feature of Dandy-Walker Syndrome. Semin Ophthalmol. 2017;32(4):501-503.
17. Ebrahimiadib N, Karkheiran S, Roohipoor R, Karkhaneh R, Modjtahedi BS. Foveal hypoplasia associated with Dandy-Walker syndrome. Can J Ophthalmol. 2017;52(4):e125-e127.
18. March WF, Chalkley TH. Sclerocornea associated with Dandy-Walker cyst. Am J Ophthalmol. 1974;78(1):54-7.
19. Nishant P, Aftab N, Raj A, Jha VC, Sinha U. Adult-onset Dandy-Walker syndrome with atypical ocular manifestations. Can J Ophthalmol. 2023;58(4):e175-e177.
20. Charan GS, Narang GS, Kaur A, Kaur E. Dandy-Walker Variant Associated with Bilateral Congenital Cataract. Int J Appl Basic Med Res. 2021;11(4):277-279.
21. de Crecchio G, Cennamo G, de Leeuw N, Ventruto ML, Lonardo MC, Friso P, et al. Severe myopia with unusual retinal anomalies and Dandy-Walker sequence in two sibs. A distinct new neuro-ocular disorder. Ophthalmic Genet. 2013;34(4):254-7.
22. Simsek T, Ozdamar Y, Simsek E, Men G. Usher syndrome associated with a variant of Dandy-Walker malformation. J Pediatr Ophthalmol Strabismus. 2010;47 Online:e1-4.
23. Ewald O, Scremin F, Busch F, Von Hertwig R. [Ocular alterations in a pediatric patient with Dandy-Walker malformations: case report]. Arq Bras Oftalmol. 2006;69(1):97-9.Portuguese.
24. Moreira LMA, Neri FB, Uzeda SQ, de Carvalho AFL, Santana GC, Souza FR, Rollemberg JC. Multiple congenital malformations including severe eye anomalies and abnormal cerebellar development with Dandy-Walker malformation in a girl with partial trisomy 3q. Ophthalmic Genet. 2005;26(1):37-43.
25. Sakurai E, Shirai S, Ozeki H, Majima A. [A case of nonrhegmatogenous retinal detachment in Dandy-Walker Syndrome]. Nippon Ganka Gakkai Zasshi. 1996;100(10):832-6.Japanese.
26. Orcutt JC, Bunt AH. Anomalous optic discs in a patient with a Dandy-Walker cyst. J Clin Neuroophthalmol. 1982;2(1):43-7.
27. Ugur HC, Torun F, Yilmaz E, Kanpolat Y. Trigeminal neuralgia in a patient with Dandy-Walker malformation. J Clin Neurosci. 2005;12(7):815-7.
28. Jha VC, Kumar R, Srivastav AK, Mehrotra A, Sahu RN. A case series of 12 patients with incidental asymptomatic Dandy-Walker syndrome and management. Childs Nerv Syst. 2012;28(6):861-7.
29. Zhang W, Chen M, Zhang W. Trigeminal neuralgia due to Dandy-Walker syndrome. J Craniofac Surg. 2013;24(4):1457-9.
30. Musa J, Rahman M, Guy A, Ahmetgjekaj I, Guy A, Kola I, et al. Trigeminal neuralgia caused by Dandy-walker malformation: A case report and systematic review of the literature. Radiol Case Rep. 2021;16(10):3084-3089.
31. Sartori MT, Bartuli A, Siboni SM, Leonardi A, Valente P, Secci J, et al. Ligneous conjunctivitis and use of human plasminogen eyedrops: The Italian experience. Haemophilia. 2023;29(2):681-684.
32. Saatci I, Yelgec S, Aydin K, Akalan N. An atretic parietal cephalocele associated with multiple intracranial and eye anomalies. Neuroradiology. 1998;40(12):812-5.
33. Behrens-Baumann W, Dust G, Rittmeier K, Langenbeck U, Vogel M. [Oculo-cerebral dysplasia: aplasia of the optic nerve with familial microphthalmos and cryptophthalmus. Clinical and computer tomography study]. Klin Monbl Augenheilkd. 1981;179(2):90-3.German.
34. Buissonniere RF, Storni V, Robain O, Ponsot G. [Joubert's syndrome]. Ann Pediatr (Paris). 1990;37(3):151-6.French.
35. Cantani A, Lucenti P, Ronzani GA, Santoro C. Joubert syndrome. Review of the fifty-three cases so far published. Ann Genet. 1990; 33(2):96-8.
36. Zhang YW, Qu HB, Long N, Leng XY, Liu YQ, Yang Y. A rare mutant of OFD1 gene responsible for Joubert syndrome with significant phenotype variation. Mol Genet Genomics. 2021;296(1):33-40.
37. Sartori S, Ludwig K, Fortuna M, Marzocchi C, Calderone M, Toldo I, et al. Dandy-Walker malformation masking the molar tooth sign: an illustrative case with magnetic resonance imaging follow-up. J Child Neurol. 2010;25(11):1419-22.
38. Assari R, Ziaee V, Moghimi S, Akbari MR, Mirmohammadsadeghi A. PHACE(S) syndrome: Report of a case with new ocular and systemic manifestations. J Curr Ophthalmol. 2016;29(2):136-138.
39. Steiner JE, McCoy GN, Hess CP, Dobyns WB, Metry DW, Drolet BA, et al. Structural malformations of the brain, eye, and pituitary gland in PHACE syndrome. Am J Med Genet A. 2018;176(1):48-55.
40. Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132(3):307-11.
41. Buzenet C, Hamel-Teillac D, Acar P, Becquet F, Curan D, Michaud V, et al. [Facial hemangioma associated with arterial anomalies, coarctation of the aorta, and eye abnormalities: PHACES syndrome]. Ann Dermatol Venereol. 2000;127(3):292-5.French.
42. Boudghene-Stambouli O, Merad-Boudia A, Abdelali S. [KID syndrome, pachydermatoglyphy and Dandy-Walker syndrome]. Ann Dermatol Venereol. 1994;121(2):99-102.French.
43. Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Lapunzina P. Oculocerebrocutaneous (Delleman) syndrome: report of two cases. Neuropediatrics. 2005;36(1):50-4.
44. Sharan S, Billson FA. Anterior megalophthalmos in a family with 3 female siblings. J Cataract Refract Surg. 2005;31(7):1433-6.
45. Sun W, Xiao X, Li S, Guo X, Zhang Q. Mutational screening of six genes in Chinese patients with congenital cataract and microcornea. Mol Vis. 2011;17:1508-13.
46. Stambolliu E, Ioakeim-Ioannidou M, Kontokostas K, Dakoutrou M, Kousoulis AA. The Most Common Comorbidities in Dandy-Walker Syndrome Patients: A Systematic Review of Case Reports. J Child Neurol. 2017;32(10):886-902.
47. Moutei H, Abdellaoui M. Sutural cataract. Pan Afr Med J. 2020;36:34.
48. Ogawa GSH. Congenital iris coloboma repair with excision of colobomatous sphincter muscle. J Cataract Refract Surg. 2021;47(8):1088-1091.
49. Meier P. [Retinal detachment in children: differential diagnosis and current therapy]. Klin Monbl Augenheilkd. 2008;225(9):779-90.German.
50. Dutton GN. Congenital disorders of the optic nerve: excavations and hypoplasia. Eye (Lond). 2004;18(11):1038-48.
51. Verdaguer P, Nadal J. Intraretinal cyst secondary to longstanding retinal detachment. Eur J Ophthalmol. 2012;22(3):506-8.
52. Hristodorov D, Lohoff T, Luneborg N, Mulder GJ, Clark SJ. Investing in vision: Innovation in retinal therapeutics and the influence on venture capital investment. Prog Retin Eye Res. 2024 Mar:99:101243.
53. Zhou ZH, Xiong PP, Sun J, Wang YL, Wang JL. Effects of posterior staphyloma on choroidal structure in myopic adults: a retrospective study. BMC Ophthalmol. 2023;23(1):406.
54. Montero LC, Belinchón I, Toledo F, Betlloch I. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27(3):162-3.
INFORMAÇÃO DOS AUTORES |
|
 |
» Francisco Victor Carvalho Barroso https://orcid.org/0000-0003-3152-8839 https://lattes.cnpq.br/8207348034186978 |
 |
» Ana Laura Eloia Limão https://orcid.org/0000-0003-4661-5660 https://lattes.cnpq.br/1401230374306671 |
 |
» Manoela Gondim https://orcid.org/0000-0001-8671-2559 https://lattes.cnpq.br/5200236785788212 |
 |
» Rian Vilar Lima https://orcid.org/0000-0003-2405-1753 https://lattes.cnpq.br/6008143139000016 |
 |
» Samuel Montenegro https://orcid.org/0000-0002-4343-7769 https://lattes.cnpq.br/7322176753684670 |
 |
» João Crispim Moraes Lima Ribeiro https://orcid.org/0000-0002-8569-1159 https://lattes.cnpq.br/5238824885154872 |
Financiamento: Declaram não haver.
Conflitos de Interesse: Declaram não haver.
Recebido em:
21 de Fevereiro de 2024.
Aceito em:
14 de Junho de 2024.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket