

Victória d’Azevedo Silveira; Thaís Saorin Conte; Patrícia Ioschpe Gus
DOI: 10.17545/eOftalmo/2021.0029
ABSTRACT
In recent decades, the advent of a wide range of sophisticated diagnostic tools and the development of gene therapies for several previously intractable conditions has promoted the growing interest in inherited eye diseases. In the current scenario, general ophthalmologists must be able to recognize the presence or association of a genetic or hereditary condition in any patient’s case and provide guidance on current diagnostic and therapeutic possibilities, but genetic counseling for patients and their families must be provided by specialists, such as geneticists or ophthalmologists qualified in genetic counseling. Therefore, the patient should be referred to a specialist.
Keywords: Cornea; Genetics; Ophthalmology.
RESUMO
Nas últimas décadas, com o advento de uma ampla gama de ferramentas diagnósticas sofisticadas e com o desenvolvimento de terapias gênicas para diversas condições antes intratáveis, interesse crescente vem sendo voltado às doenças oculares herdadas. No atual cenário, o oftalmologista geral deve ser capaz de reconhecer a presença ou associação de uma condição genética ou hereditária no caso de algum paciente e fornecer orientação quanto às possibilidades diagnósticas e terapêuticas vigentes, porém o aconselhamento genético ao paciente e/ou família deve ser fornecido por especialistas, como geneticistas e/ou oftalmologistas com título em aconselhamento genético, portanto realizar o encaminhamento para este fim.
Palavras-chave: Córnea; Genética; Oftalmologia.
INTRODUCTION
The Porto Alegre University Clinics Hospital (HCPA) is a reference in inherited metabolic diseases throughout Brazil, standing out in the collaboration of its experiences and care throughout Latin America. Although not very prevalent individually (around 1:100,000 births), they occur in 1:800 live births when examined collectively. This prevalence is similar to that of Down syndrome, the most common chromosomal syndrome, which is highly relevant for public health1-3.
The HCPA Ophthalmology Clinic has a significant interface with the hospital’s Medical Genetics Clinic. The former clinic cares for the latter’s patients to aid in cases of uncertain diagnoses, to support adherence and effectiveness of treatments, and to diagnose ocular complications. Likewise, the HCPA Ophthalmology Clinic is a reference in corneal transplantation in the Brazilian state of Rio Grande do Sul, where the clinic is located, allowing the most varied corneal pathologies to be cared for.
This article intends to contribute to the reader’s knowledge about the HCPA Ophthalmology Clinic’s experience with inherited corneal diseases, whether they are consequences of systemic genetic diseases or primarily ocular but with a marked genetically inherited character. The most prevalent of these diseases will be addressed in this article.
Conditions with Primarily Corneal Involvement:
1.1 Dystrophies
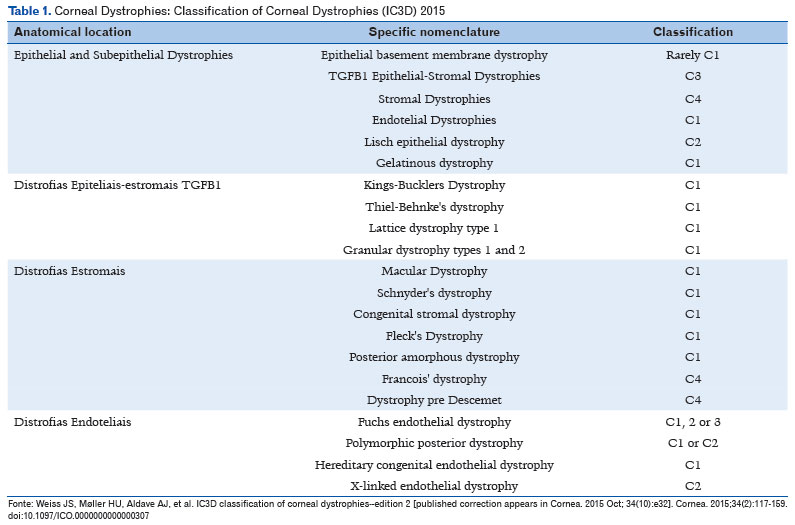
Corneal dystrophies are a group of bilateral, symmetrical, slowly progressive diseases whose evolution is independent of environmental interference. They are traditionally classified according to the corneal structure primarily affected, although more than one layer may be involved during their progression as follows: epithelial, subepithelial, Bowman’s layer, stromal, Descemet’s, and endothelial1-3.
Genotype analyses have been changing concepts and introducing evidence of heterogeneous and variable genetic and phenotypic expression. For example, some dystrophies can be unilateral, whereas others may be asymmetric, and others continue to lack laboratory evidence. Within this new paradigm, variations in a single gene can lead to different phenotypes, as in the case of the TGFBI gene, and some dystrophies can be caused by variations in different genes, as occurs with Meesmann corneal dystrophy1-3.
In 2008, the Cornea Society established a new classification for corneal dystrophies, based on anatomical criteria and grouping them into subgroups according to a common genetic basis. Corneal dystrophies were divided into the following categories:
• Category 1: gene mapped, and specific mutations identified.
• Category 2: one or more chromosomal loci mapped, but the gene needs to be identified.
• Category 3: a chromosomal locus has not yet been identified.
• Category 4: either a new or suspicious dystrophy or a known one, but with weak evidence.
In 2015, this classification underwent some changes in terms of anatomical classification. Dystrophies had been previously classified according to the most affected corneal lamella. However, eventually it was realized that it was more important to determine the primary (originating) type of cell, since corneal dystrophies usually affect more than one lamella.
The current classification of dystrophies is summarized in Table 1. This article will not describe every one of the dystrophies, given the complexity of the subject, which would require a separate chapter3.
1.2 Corneal Ectasias
Keratoconus is an asymmetric, progressive, usually bilateral corneal ectasia that is clinically characterized by corneal thinning and asymmetric astigmatism, with an onset usually in adolescence. It is considered multifactorial, having both genetic and environmental influence in its development. Its reported prevalence has been increasing with the possibility of early diagnosis, i.e., it can vary from 1:50 in areas of frequent consanguineous marriages and hot and dry climate, such as India and Iran, to 1:300 in areas previously considered to be of low risk and prevalence, such as The Netherlands. There seems to be a strong correlation between the appearance of keratoconus and the habit of scratching the eyes, as in allergic patients4-10.
In large population studies, family history varies between 5% and 20%, with a mean of 12%-13%4-11. Keratoconus is considered a heterogeneous genetic condition, with incomplete penetrance and variable expression. Most familial cases are of autosomal dominant inheritance with variable expression. Many genes are involved, including homeobox VSX1, SOD1, TGFB1, MIR 184, COL4A3/COL4A4, and FLG. The association of keratoconus with other ocular and systemic diseases is not uncommon (Table 2)4-10.
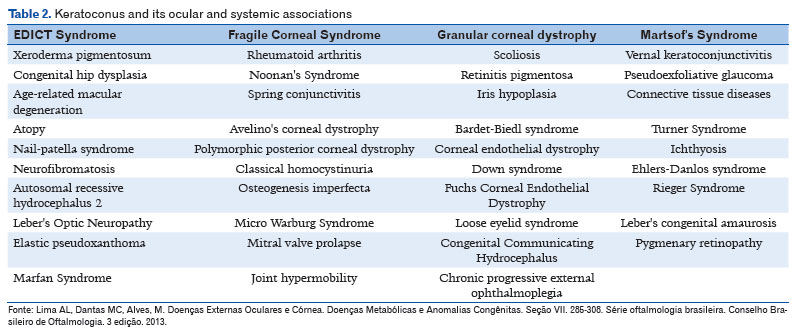
Currently, evidence of a genetic component recommends more careful attention to the family members of patients affected by the disease. Tomographic and topographic studies of the cornea of relatives are useful to identify subclinical cases and enable an early diagnosis and effective treatment if clinical worsening occurs4-10.
Posterior Keratoconus
Posterior keratoconus is characterized by an indentation in the central or paracentral region of the posterior cornea, without the protrusion of the anterior surface of the cornea that is typical of keratoconus. It is classified as a congenital anomaly as it is associated with an anterior displacement of Descemet’s membrane that is present since birth. Posterior keratoconus is usually stable; however, it can cause irregular astigmatism and amblyopia. Most cases are unilateral and sporadic11.
1.3 Megalocornea
Megalocornea is characterized by a horizontal diameter of the cornea measuring 13 mm or more. It is bilateral, congenital, and non-progressive. About 90% of affected patients are male12,13.
It has been suggested that megalocornea occurs due to changes in collagen production, as it may be associated with Marfan syndrome. It can also be associated with goniodysgenesis, cataract, ectopia lentis, and congenital glaucoma, among other conditions. Megalocornea is typically an X-linked autosomal recessive condition owing to a mutation in the CHRDL1 gene. However, autosomal dominant inheritance has been reported12,13.
Differential diagnosis with congenital glaucoma is made by measuring intraocular pressure and by biomicroscopy and optic-nerve findings. Ultrasound can also be used to assess axial length, which in megalocornea cases will be normal12,13.
1.4 Microcornea
Microcornea is characterized by a corneal diameter smaller than 10 mm in adults or smaller than 9 mm in newborns. It can be either unilateral or bilateral. It occurs due to a mutation in the PAX6 gene, whose inheritance can be either autosomal dominant or recessive. Approximately 20% of patients develop glaucoma over their lifetime. Microcornea can occur either alone or in association with other ocular conditions, such as congenital cataract and dysgenesis of the anterior segment. It can also be associated with systemic conditions, such as fetal alcohol syndrome, achondroplasia, and Ehler-Danlos syndrome12,13.
1.5 Cornea Plana
Cornea plana is characterized by a radius of corneal curvature less than 43 diopters. A pathognomonic finding is the curvature of the cornea being equal to that of the sclera. It occurs due to a mutation in the KERA gene, whose inheritance can be either autosomal dominant or recessive12,13. This gene encodes proteoglycans that are important for the disposition of collagen fibrils in the cornea.
Cornea plana is usually associated with high degrees of hyperopia (above 10 diopters) and a shallow anterior chamber, predisposing to closed-angle glaucoma. It may either be an isolated finding or occur as a part of Ehler-Danlos syndrome12,13.
1.6 Posterior Embryotoxon
Posterior embryotoxon is characterized by a thickened and anteriorly displaced Schwalbe line (at the junction between the trabeculae and the edge of Descemet’s membrane), which can be observed on ectoscopy. It is usually a bilateral and dominantly inherited finding, either isolated or associated with other anomalies of the anterior segment, including Axenfeld-Rieger syndrome12,13.
1.7 Axenfeld-Rieger Syndrome
Axenfeld-Rieger syndrome is a spectrum of conditions caused by the anomalous development of anterior-segment structures derived from the neural crest. It can occur simultaneously with microcornea, posterior embryotoxon, hypoplasia of the iris, corectopia, uveal ectropion, glaucoma, and other changes. This syndrome is bilateral, has no predilections for either sex, has an autosomal dominant inheritance in 75% of cases, and can also be sporadic. It can be caused by mutations in the PAX6, FOXC1, or PITX2 genes. It may or may not be associated with systemic alterations, including maxillary hypoplasia, hypertelorism, dental abnormalities, and others12,13.
1.8 Peters Anomaly
Peters anomaly is characterized by the absence of Descemet’s membrane and endothelium in a portion of the cornea, causing central or paracentral opacity from birth. It is caused by a defect in the migration of cells from the neural crest, and is usually bilateral. Most cases occur sporadically, but the anomaly can also occur owing to autosomal dominant or recessive inheritance. Half of the patients develop glaucoma from associated angular anomalies. Peters anomaly is divided into the following categories:
- Type I: This type is associated with occurrence of iridocorneal adhesion. Opacity is usually avascular, either central or total. This type is caused by mutations of the PITX2, FOXC1, CYP1B1, and PAX6 genes, and it has the best prognosis.
- Type II: This type is considered with the occurrence of corneolenticular adhesion associated with cataract. Opacity is usually vascularized and may be either central or total. This type occurs due to a mutation of the FOXE3 gene.
- “Peters plus” syndrome: This category is considered when the syndrome is associated with systemic conditions, including short stature, cleft palate, cardiac alterations, and others12,13.
1.9 Sclerocornea
Sclerocornea is a non-progressive, non-inflammatory condition, in which the cornea becomes a continuation of the sclera. It can be limited to the periphery or affect the entire area of the cornea. It tends to be sporadic, but it can also be recessive or dominant12,13.
1.10 Congenital Dermoid
Congenital dermoid is a solid benign tumor that can be either unilateral or bilateral, with little or no growth. It is considered a choristoma, as it contains cellular elements that are not usually present on the ocular surface, such as hair follicles and sebaceous glands. Unilateral cases are sporadic and predominantly superficial, whereas bilateral cases are associated with Goldenhar syndrome, which is characterized by congenital dermoid, auricular appendices, and vertebral abnormalities. In Goldenhar syndrome, dermoids may extend down to the deep corneal stroma, or even the entire extension of the anterior segment may be affected12,13.
Conditions Secondary to Systemic Involvement:
Several systemic conditions can lead to accumulation of substances in the cornea, affecting its transparency. When these corneal conditions are first identified by the ophthalmologist, this may contribute to the diagnosis of the systemic condition and thereby save lives12.
2.1 Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) are a group of lysosomal diseases caused by a deficiency of enzymes that metabolize glycosaminoglycans, resulting in an accumulation of these substances and in the impairment of cell metabolism in multiple systems. MPS have extensive phenotypic variation and are classified into 6 groups. In most cases, the pattern of inheritance is autosomal recessive, except in type II MPS, which is X-linked12,15,16.
The phenotypic presentation may include rough facial features, short stature, enlarged organs, skeletal dysplasia, cardiac, pulmonary, and ophthalmological disease, mental deficiency, and behavioral changes, all in varying degrees12,15,16.
It is extremely important for the ophthalmologist to have already seen an image of these patients, as this is the only way a diagnosis of MPS may be considered. The phenotype worsens over time, and some patients die young. Ocular changes are varied, and may include high hyperopia, corneal opacity, scleral thickening, glaucoma and pseudoglaucoma, retinal dystrophy, and optic atrophy12,15,16.
A grayish and homogeneous opacity in varying degrees is observed on biomicroscopy. Transparency is lost in a diffuse, bilateral, and slowly progressive manner, evenly affecting the cornea, from one limbus to the other and thereby making the cornea more rigid. This consequently increases intraocular pressure and favors the development of hyperopia. An increase in the volume of keratocytes and changes in collagen stromal fibrils are observed in the histopathological examination12,15,16.
Turbidity is a significant cause of photophobia and loss of corrected visual acuity. It is observed in the following MPS subgroups: MPS IH (Hurler), MPS IS (Scheie), MPS IH/S (Hurler/Scheie), MPS IV (Morquio), and MPS VI (Maroteaux-Lamy). Corneal turbidity is more frequent in MPS IH and even in attenuated MPS I phenotypes, being found in 70% of patients12,15,16.
A cooperative study that evaluated transplants performed in these patients confirmed that intraocular pressure returns to normal values in most cases in which corneas were transplanted, and reported improved vision and persistence of graft transparency. These results have a positive impact on patient quality of life. However, systemic enzyme replacement therapy may not prevent corneal opacification16.
2.2 Fabry Disease
Fabry disease is a congenital defect in glycosphingolipid metabolism that is secondary to deficient α-galactosidase activity. It presents an X-linked inheritance pattern. More than 200 mutations of the GLA gene, which encodes α-galactosidase A, have been described12,17,19.
Ocular findings play an important role in Fabry disease, as they are one of the earliest clinical manifestations of the disease and usually appear in the second decade of life. Cornea verticillata occurs as the result of deposits in the basal laminae of the epithelium. Other ocular findings include cataract, conjunctival aneurysm, and papillary edema, among others12,17,19.
The onset and progression of Fabry disease manifestations are highly variable. Patients with more aggressive forms often present with severe peripheral pain and progress to multiple organ failure in the first decade of life. In addition to ocular changes, there may be acroparesthesia, angiokeratomas, hypohidrosis, deafness, kidney failure, and ischemic cardiac and cerebral complications12,17,19.
Behavioral problems can be the first sign in adolescents and adults, who are sometimes first diagnosed as psychiatric patients12,17,19.
Cystinosis
Cystinosis is a condition characterized by the accumulation of cystine crystals, particularly in the eyes and kidneys, owing to a defect in the CTNS gene. It is a rare metabolic disorder with autosomal recessive inheritance. It is classified into the infantile, intermediate, and juvenile forms. The infantile form is severe and causes kidney failure and death in the first decade of life. The cystine crystals cause intense photosensitivity, but otherwise do not usually significantly lower visual acuity. The treatment of ocular manifestations is performed with cysteamine eye drops in multiple daily applications, if possible as many as ten12,19.
Wilson Disease
Wilson disease is characterized by copper deposition in the liver, and in some cases in the brain and in Descemet’s membrane, which are usually simultaneously affected. This is an autosomal recessive condition that affects the ATP7B gene. The classic sign is the mostly golden-brown peripheral Kayser-Fleischer ring in Descemet’s membrane. Liver transplantation may be needed to control the disease. The corneal findings are useful to monitor the therapeutic response, as they tend to disappear as the disease is controlled12,19.
DISCUSSION
Currently, owing to the rapid advances in the field of medical genetics , new paradigms and concepts emerge. This has allowed researchers to better understand diseases, change prognoses, and genetic counseling for countless patients and their families.
A common doubt is whether there is a need to request for genetic testing and whether such tests are really beneficial in primarily corneal diseases. Genetic tests must be requested and interpreted by experts or specialists in the field, as the results are relatively broad, can provide unexpected information, and can cause anxiety. Therefore, when genetic tests are indicated, it is suggested that the patient be referred to a genetics specialist to assess whether the test is indicated or necessary. It must always be made sure that the patient seeks a genetics professional first, rather than a laboratory directly, and that the patient always receives a copy of their test, if it is performed20.
From an ophthalmological point of view, diagnosis and treatment continue to be based on clinical criteria, despite recent advances in gene therapy for retinal dystrophies caused by the RPE65 gene. The future rests on the possibility of gene therapy with the introduction of DNA sequences with a therapeutic effect tailored to each clinical condition, allowing the correction of very specific gene defects in monogenic diseases, or at least minimizing damage in polygenic diseases20.
Awareness of these diseases by ophthalmologists is extremely relevant, considering that the patients are likely to consult an ophthalmologist before any other physician. Many genetic diseases can be treated, and the best results for preventing complications occur when treatment is started early. This can prevent the appearance or progression of ocular and systemic defects, and can protect the lives of patients.
Although gene therapies are not yet available for the vast majority of ocular genetic conditions, diagnostic investigation is the best indication. At HCPA, suspected cases of syndromes that progress with systemic involvement are immediately referred to the Medical Genetics Clinic, whereas cases that progress exclusively with ocular involvement are diagnosed and followed up by an ophthalmologist. There are specialized laboratories that perform diagnostic panels encompassing different diseases. Some of the diseases for which HCPA offers genetic testing include Bietti crystalline corneoretinal dystrophy, the microphthalmia spectrum, anophthalmia, coloboma, Alagille syndrome, mucopolysaccharidosis type IIIC, Stickler syndrome, and many others.
REFERENCES
1. Klintworth GK. Genetic disorders of the cornea: from research to practical diagnostic testing. Clin Exp Ophthalmol. 2005;33(3):231-2.
2. Vincent AL, Patel DV, McGhee CN. Inherited corneal disease: the evolving molecular, genetic and imaging revolution. Clin Exp Ophthalmol. 2005;33(3):303-16.
3. Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, et al. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015;34(2):117-59.
4. Godefrooij DA, de Wit GA, Uiterwaal CS, Imhof SM, Wisse RP. Age-specific Incidence and Prevalence of Keratoconus: A Nationwide Registration Study. Am J Ophthalmol. 2017 Mar;175:169-72.
5. Shetty R, Kaweri L, Pahuja N, Nagaraja H, Wadia K, Jayadev C, et al. Current review and a simplified “five-point management algorithm” for keratoconus. Indian J Ophthalmol. 2015;63(1):46-53.
6. Hashemi H, Heydarian S, Yekta A, Ostadimoghaddam H, Aghamirsalim M, Derakhshan A, et al. High prevalence and familial aggregation of keratoconus in an Iranian rural population: a population-based study. Ophthalmic Physiol Opt. 2018;38(4):447-55.
7. Hashemi H, Heydarian S, Hooshmand E, Saatchi M, Yekta A, Aghamirsalim M, et al. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea. 2020;39(2):263-70.
8. Godefrooij DA, de Wit GA, Uiterwaal CS, Imhof SM, Wisse RP. Age-specific Incidence and Prevalence of Keratoconus: A Nationwide Registration Study. Am J Ophthalmol. 2017 Mar;175:169-72.
9. Shetty R, Kaweri L, Pahuja N, Nagaraja H, Wadia K, Jayadev C, et al. Current review and a simplified “five-point management algorithm” for keratoconus. Indian J Ophthalmol. 2015;63(1):46-53.
10. Hashemi H, Heydarian S, Yekta A, Ostadimoghaddam H, Aghamirsalim M, Derakhshan A, et al. High prevalence and familial aggregation of keratoconus in an Iranian rural population: a population-based study. Ophthalmic Physiol Opt. 2018;38(4):447-55.
11. Gus PI, Araújo BS, Zelanis S, Schmalfuss TR, Marinho DR. Posterior keratoconus and iris atrophy: a fortuitous association? Arq Bras Oftalmol. 2019;82(1):68-71.
12. Lima AL, Dantas MC, Alves, M. Doenças Externas Oculares e Córnea. Doenças Metabólicas e Anomalias Congênitas. Seção VII. 285-308. Série oftalmologia brasileira. Conselho Brasileiro de Oftalmologia. 3 edição. 2013.
13. Dantas AM, Sallum JMF. Embriologia, genética e malformações do aparelho visual. Série oftalmologia brasileira. Conselho Brasileiro de Oftalmologia. 3 edição. 2013.
14. Del Longo A, Piozzi E, Schweizer F. Ocular features in mucopolysaccharidosis: diagnosis and treatment. Ital J Pediatr. 2018;44(Suppl 2);125.
15. Villas-Bôas FS, Fernandes Filho DJ, Acosta AX. Achados oculares em pacientes com mucopolissacaridoses. Arq Bras Oftalmol. 2011;74(6):430-4.
16. Ohden KL, Pitz S, Ashworth J, Magalhães A, Marinho DR, Lindahl P, et al. Outcomes of keratoplasty in the mucopolysaccharidoses: an international perspective. Br J Ophthalmol. 2017;101(7):909-12.
17. Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A. Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Exp Ophthalmol. 2005;33(2):164-8.
18. Cordeiro CA, Oréfice F, Lasmar EP, Santos HH, Valadares ER. Córnea verticilata - marcador clínico da doença de Fabry: relato de caso. Arq Bras Oftalmol. 2007;70(4):701-5.
19. Poll-The BT, Wenniger-Prick CJMB. The eye in metabolic diseases: clues to diagnosis. Eur J Paediatr Neurol. 2011;15(3):197-204.
20. Recommendations for Genetic Testing of Inherited Eye Diseases. American Academy of Ophthalmology. 2014. Available at: https://www.aao.org/clinical-statement/recommendations-genetic-testing-of-inherited-eye-d
AUTHOR’S INFORMATION
Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
July 21, 2020.
Accepted on:
December 15, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket