

Samilla Augusto Vieira de Araujo1; Renata Zaltron Neumann1; Vitor Gilberto Essi Monticuco2; Breno Reis Almeida2; Francyne Veiga Reis Cyrino3
DOI: 10.17545/eOftalmo/2021.0026
RESUMO
A Amaurose Congênita de Leber (ACL) é uma distrofia retiniana hereditária de início precoce, do nascimento ao 1º ano de vida, caracterizada por deficiência visual severa. Atualmente estão identificados 14 genes cujas mutações se associam a este fenótipo e a transmissão é de modo autossômico recessiva. Relatamos um paciente do sexo masculino de 17 anos com queixa de baixa acuidade visual (BAV) severa desde a infância em ambos os olhos, sem diagnóstico e sem antecedentes familiares de baixa acuidade visual severa. Da história familiar destaca-se o fato dos pais serem primos consangüíneos de primeiro grau. Foi realizado angiofluoresceinografia (AGF), tomografia de coerência óptica (OCT), eletrorretinograma (ERG) e teste genético, confirmando o diagnóstico de amaurose congênita de Leber. Atualmente existem estudos com reposição de terapia genética com resultados promissores, mas que ainda não contemplam todas as variantes.
Palavras-chave: Distrofias da Retina; Amaurose; Distrofias Congênitas; Degeneração da Retina.
ABSTRACT
Leber congenital amaurosis (LCA) is an early-onset hereditary retinal dystrophy that becomes apparent in the first year of life and is characterized by severe visual impairment. Currently, 14 gene mutations that are associated with this phenotype have been identified and the transmission is autosomal recessive. We report a case of a 17-year-old male patient complaining of severe low visual acuity in both eyes since childhood, but a diagnosis had not been made. The patient did not have a family history of severe visual acuity. The family history was revealed that his parents were first cousins (consanguineous union). Fluorescein angiography, optical coherence tomography, electroretinography, and genetic testing were performed, which confirmed LCA diagnosis. Currently, studies on gene replacement therapy with promising results exist; however, they do not yet include all variants.
Keywords: Retinal Dystrophies; Amaurosis; Congenital Dystrophies; Retinal Degeneration.
INTRODUÇÃO
A amaurose congênita de Leber é a designação para um grupo de distrofias retinianas de acometimento precoce, de caráter hereditário, caracterizada por deficiência visual moderada a severa, identificada nos primeiros meses de vida. A acuidade visual quando pode ser avaliada varia de 20/200 a ausência de percepção luminosa, raramente é melhor que 20/2001.
Na maior parte dos casos a transmissão é de modo autossômico recessivo, apesar de existirem alguns casos descritos com transmissão dominante. Atualmente estão identificados 14 genes cujas mutações se associam a este fenótipo.
Durante os primeiros meses de vida os pacientes desenvolvem nistagmo, os reflexos fotomotores frequentemente encontram-se diminuídos ou abolidos, alta hipermetropia, enoftalmia, sendo comum o sinal oculodigital de Franceschetti. Posteriormente podem aparecer catarata e ceratocone2.
Alguns critérios são utilizados nos trabalhos publicados para caracterizar os pacientes com ACL:
- Cegueira ou BAV severa nos seis primeiros meses de vida ou no 1° ano de vida6,10.
- ERG extinto ou muito reduzido (fotópico e escotópico)2,3,4,10.
- Fundus de aspecto normal ou com alterações pigmentares minimas3 e/ou estreitamento arteriolar modesto2,3.
- Ausência de outras desordens multissistêmicas ou retinianas4,10.
Os achados de fundo de olho nos primeiros meses de vida são normais ou alterações mínimas, dificultando o diagnóstico. O exame de ERG é a chave para estabelecer o diagnostico2, onde se detecta pouca ou nenhuma atividade na retina, indicando baixa função visual. Com o tempo é comum o aparecimento de outras alterações na retina, como atenuação vascular, atrofia nervo óptico (principalmente após primeiro ano), alterações pigmentares tipo espículas ósseas, acometimento macular tipo granular ou colobomatoso, lesões numulares pigmentadas no polo posterior e periferia3.
Em alguns casos a ACL é relacionada com complicações do sistema nervoso central, tais como atraso do desenvolvimento, epilepsia e deficiência de habilidades motoras. Como a ACL é relativamente rara, a freqüência das complicações do sistema nervoso central são ainda desconhecidas3.
O diagnóstico diferencial deve ser feito com cegueira noturna estacionária congênita, acromatopsia, retinose pigmentar infantil, síndrome de Joubert, Síndrome de Zellweger e doença de Refsum infantil6.
RELATO DE CASO
Paciente E.F.D., 17 anos masculino, estudante, apresentava queixa de baixa acuidade visual desde a primeira infância em ambos os olhos (AO). Relatava movimento involuntário dos olhos (nistagmo) de início aos 6 meses de idade quando foi avaliado e aventada a hipótese diagnóstica de Amaurose congênita da Leber, sem porém ter sido realizado outros exames complementares além da fundoscopia (SIC). Referia fotofobia, dificuldade visual na escola e algum grau de déficit de aprendizado, usando impressos ampliados e por vezes relata apertar o olho com o dedo para enxergar melhor (SIC). Paciente sem antecedentes familiares de cegueira ou baixa acuidade visual. Ao exame, observa-se nistagmo horizontal bilateral, acuidade visual corrigida de 20/200 em ambos os olhos (OD: +2,50 esf = - 0,75 cil 900 e OE:+3,50 esf = - 1.00 cil 750), biomicroscopia sem anormalidades, tonometria: 10mmHg em AO. À fundoscopia de AO, observado coloboma macular e áreas de hiperplasia do epitélio pigmentado na área do coloboma assim como na periferia dos quatro quadrantes. (Figura 1).
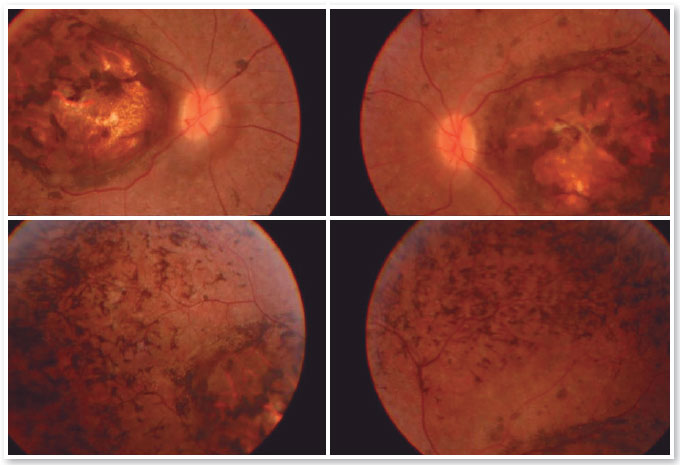
Os exames complementares para diagnóstico foram solicitados e estão relacionados nas figuras abaixo.
DISCUSSÃO
A amaurose congênita de Leber é a mais grave de todas as distrofias hereditárias da infância e sua principal característica é a baixa acuidade visual severa ao nascimento ou no primeiro ano de vida. Geralmente o diagnóstico é feito ao redor dos 2 anos de idade, época em que os pais observam os sintomas dos pacientes, como o foi do caso descrito.
Todos os pacientes com ACL são classificados como portadores de visão subnormal profunda ou quase cegueira. A baixa acuidade visual severa é relatada em todos os trabalhos da literatura. O erro refracional mais comum é a hipermetropia13 como observado neste caso. Os principais sintomas são compressão óculo-digital, enoftalmo, estrabismo, nistagmo e fotofobia1,5,6,10.
A fundoscopia tem sido descrita como normal ou minimamente alterada nos estágios iniciais, havendo posterior desenvolvimento de alterações de aspecto extremamente variável. O principal exame para o diagnóstico precoce da ACL é o ERG, que se apresenta extinto ou quase extinto, mesmo sem alterações de fundo de olho.
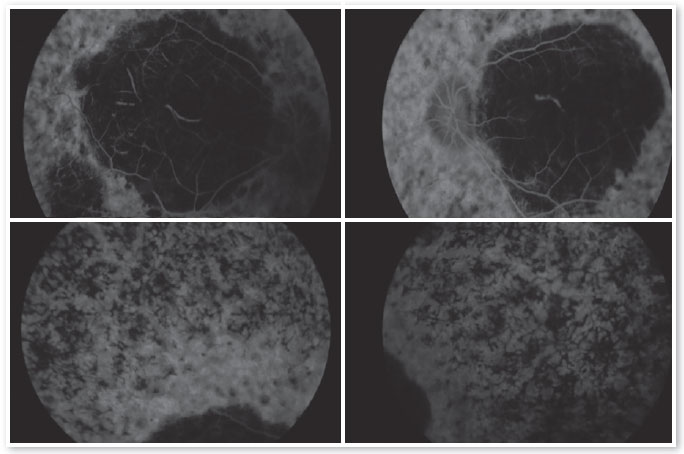
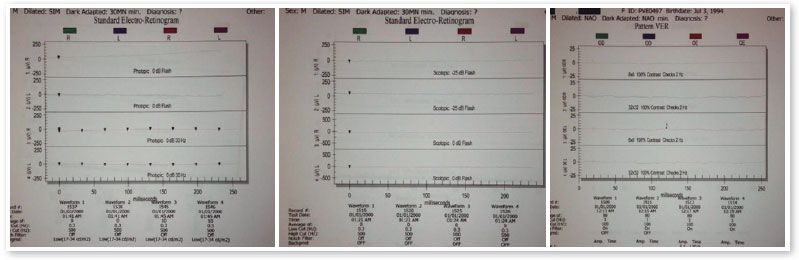
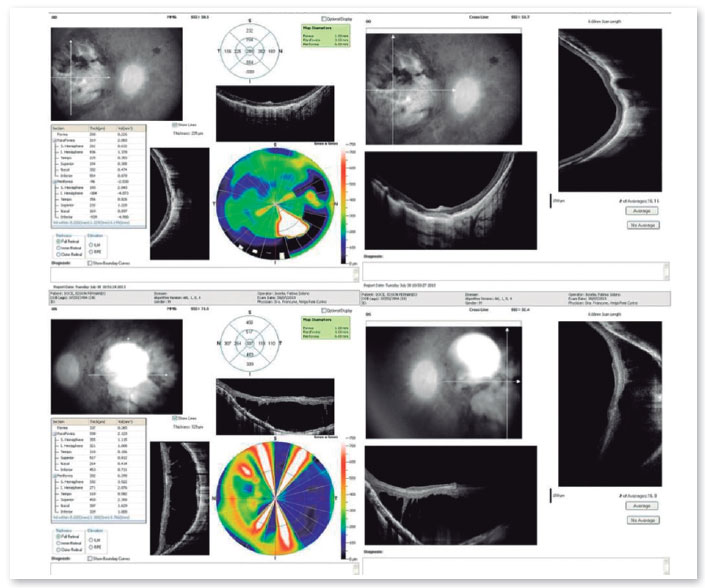
O retardo do desenvolvimento neuropsicomotor associado à ACL tem sido questionado, podendo decorrer apenas de privação sensorial imposta pela doença.
Vem sendo estudado a anos uma proposta de terapia de reposição genética para ACL. Recentemente o Voretigenene parvovec-rzyl(Luxturna®) foi aprovado pelo FDA sendo indicado para crianças e adultos com casos confirmados de distrofia retinal provocada por mutações bialélicas do gene RPE65, ou seja, em ambas as cópias do DNA herdadas do pai e da mãe. A terapia funciona por meio da introdução de uma cópia normal do gene em células da retina com a ajuda de adenovírus que a levam ao núcleo das células defeituosas. Isso permite que as células passem a produzir a proteína que converte a luz em um sinal elétrico que pode ser transmitido pelo nervo ótico, efetivamente restaurando a visão15,16.
A terapia é aplicada apenas uma vez em cada olho, com pelo menos seis dias entre os procedimentos cirúrgicos. As reações adversas mais comuns do tratamento incluíram hiperemia conjuntival, catarata e aumento da pressão intraocular. Acredita-se que nos Estados Unidos, cerca de 1.000 a 2.000 pessoas apresentem a mutação15,16.
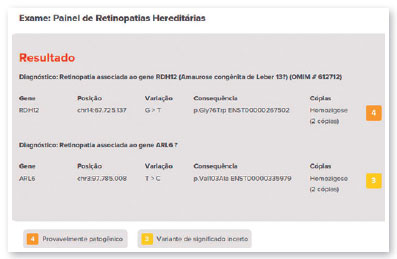
Neste caso, o teste genético não apresentou a mutação ligada ao gene RPE65. A variante detectada foi associada ao gene RDH12, uma variante envolvida com a redução de all-trans-retinal e de seus isômeros (PMID:15258582), neste caso herdadas devido a consangüinidade dos pais (pais primos em primeiro grau, variante familiar).
Nos casos de distrofias hereditárias, se faz mandatório o aconselhamento genético para reduzir a possibilidade de transmissão nas próximas gerações. No caso apresentado, se o paciente não se casar dentro da família, a mutação não será transmitida.
Contudo, infelizmente, até o momento, não há tratamento para esta variante, mas estudos estão em andamento e, em um futuro não muito distante, espera-se que seja possível reduzir e até mesmo tratar a progressão destas condições tão debilitantes.
AGRADECIMENTOS
Á Dra Juliana Salum que foi a intermediadora para realização do teste genético, fornecido gratuitamente pela empresa Mendelics. Ao Dr. Mario Ogata, pelo referenciamento do paciente e seguimento.
REFERÊNCIAS
1. Steinberg A, Ronen S, Zlotogorski Z, Silverston BZ, Hirsch I, Nawratzki. J PediatrOphthalmolStrabismus1992;29:2
2. Schroeder R, Mets MB,Maumenee IH; Leber’s congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. ArchOphthalmol 1987;105(3):356-9.
3. Leber congenital amaurosis – researchadvances - http://www.blindness.org/leber-congenital-amaurosis acessado em 19/04/2018
4. Vaizey MJ, Sanders MD, Wybar KC, Wilson J. N. Neurological abnormalities in congenital amaurosis of Leber – 1977
5. Nickel B, Hoyt CS. Leber’s congenital amaurosis - is mental retardation a frequent associated defect? - 1982
6. Steinberg A, Ronen S, Zlotogorski Z, Silverston BZ, Hirsch I, Nawratzki. J PediatrOphthalmol Strabismus 1992;29:224-227
7. Lambert SR, Taylor D, Kriss A. The infant with nystagmus, normal appearing fundi, but an abnormal ERG. SurvOphthalmol 1989a;34(3):173-186.
8. Casteels I, Spileers W, Demaerel P, Casaer P. De Cook P., Dralands L, Missotten L. Neuropediatrics 1996; 27:183-193
9. Noble KG, Carr RE. Leber congenital amaurosis. A retrospective study of 33 cases and an histopatological study of one case. ArchOphthalmol 1978; 96:818-821
10. Schroeder R, Mets MB,Maumenee IH; Leber’s congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. ArchOphthalmol 1987;105(3):356-9.
11. Dekaban A. Hereditary syndrome of congenital night blindness (Leber), polycystic kidneys and maldevelopment of the brain. AM. J. Ophthalmol1969;68:1029-1037. Apud c Nnickel e Hoyt.
12. Fulton AB, Hansen RM, Mayer L; Vision in Leber Congenital Amaurosis. ArchOphthalmol 1996; 114 (6): 698-703.
13. Lambert SR, Kriss A, Taylor D, Coffrey R, Pembey M. Follow up and diagnostic reappraisal of 75 patients with Leber’s congenital amaurosis. 1989b
14. Fishman GA. Electrophysiologic testing in disorders of the retina, optic nerve and visual pathway. Ophthalmology Monographs. The Foundation of the American Academy of Ophthlamology. 2001.308p.
16. Luxturna [prescribing information]. Philadelphia, PA; Spark Therapuetics, Inc. December 2017.
INFORMAÇÃO DOS AUTORES




*Residentes do Centro Avançado em Oftalmologia- Universidade de Ribeirão Preto – CAO-UNAERP.
** Médico oftalmologista.
** Preceptora da residência de oftalmologia do CAO-UNAERP, medica assistente do HCFMRP-USP Ribeirão.
Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
21 de Julho de 2020.
Aceito em:
15 de Dezembro de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket