

Ingrid de Oliveira Haubert Magalhães1; Priscila Hae Rim2; Marcelo Torigoe3
DOI: 10.17545/eOftalmo/2020.0017
ABSTRACT
Eyeball malformations are caused by defects or ruptures at the stage of differentiation and/or development of specialized tissues and cells. This article mainly addresses three congenital eye anomalies, i.e., microphthalmia, anophthalmia, and coloboma, which are congenital, structural eye malformations that can occur in isolation or as part of a syndrome (associated with craniofacial, skeletal, genitourinary, cerebral, or cardiac anomalies, among others). Considering the substantial impact of eyeball malformations on the lives of children and their families, we conducted a narrative literature review on the subject, including its main classifications, related genes, and current perspectives, using articles on microphthalmia, anophthalmia, and coloboma retrieved from the Bireme/PubMed database. Based on the studies that have been conducted, the possibility of using novel treatment approaches at an early stage of fetal eye development in the future is expected, preventing genetic alterations or failures during this vital period of eye formation.
Keywords: Coloboma; Microphthalmia; Anophthalmia; Eye abnormalities.
RESUMO
As malformações do globo ocular são ocasionadas por defeito ou ruptura na fase de diferenciação e/ou desenvolvimento de tecidos e células especializadas. Neste artigo são abordadas principalmente três anomalias congênitas dos olhos, ou seja, microftalmia, anoftalmia e coloboma (MAC), que são malformações oculares estruturais congênitas que podem ser isoladas ou fazer parte de uma síndrome (associadas a anomalias craniofaciais, esqueléticas, genitourinárias, cerebrais, cardíacas, entre outras). Considerando o impacto importante das malformações do globo ocular na vida das crianças e de seus familiares, os autores realizaram uma revisão narrativa da literatura sobre o tema, incluindo suas principais classificações, os genes relacionados e as perspectivas atuais. Foram pesquisados na base de dados Bireme/Pubmed artigos sobre microftalmia, anoftalmia e coloboma. A partir dos estudos que vêm sendo realizados, espera-se que no futuro seja possível realizar novas abordagens terapêuticas durante uma fase precoce do desenvolvimento ocular no feto para evitar que alterações genéticas ou falhas ocorram nesse período importante da formação ocular.
Palavras-chave: Coloboma; Microftalmia; Anoftalmia; Anormalidades do olho.
INTRODUCTION
The worldwide incidence of microphthalmia, anophthalmia, and coloboma (MAC) was estimated to be 6-13 per 100,000 live births; microphthalmia at 2-17 per 100,000, anophthalmia at 0.6-4.2 per 100,000, and coloboma at 2-14 per 100,000 live births (1).
Eyeball malformations are caused by ruptures or defects during the development and/or differentiation of specialized tissues and cells (2). Most cause significantly low vision during childhood (3,4) and have a major impact on the lives of children and their families.
In this article, three major congenital anomalies of the eyes will be addressed: microphthalmia, anophthalmia, and coloboma. MAC are congenital, structural eye malformations that can occur in isolation or as part of a syndrome (3,5,6). The incidence of other associated anomalies varies among studies, from one third (1) to 90% (7). When the condition is syndromal, it may present craniofacial, skeletal, renal, genital, cardiac, cerebral, and other anomalies. Neurological alterations are more commonly associated with anophthalmia, whereas urological and genital defects are more prevalent in patients with coloboma and microphthalmia (1).
The objective of this review is to better understand existing classifications and retrieve information on genetic mapping, early treatment, and improved patient follow up.
METHODS
This narrative literature review was conducted using the Bireme/PubMed database and comprises articles on MAC. The search strategy included the years from 1950 to 2020 and the following keywords: “microphthalmia,” “anophthalmia,” “coloboma,” “congenital eye malformations,” “eyeball anomaly,” and “MAC,” in addition to their different combinations. A total of 102 articles were found and 33 of them were selected, which featured MAC data, including definition, embryogenesis, classifications, and associated syndromes and genes. Case reports, experimental or animal studies, and studies that were not statistically significant were excluded.
RESULTS
Microphthalmia is defined as a condition wherein the total axial length (TAL) of the eye is less than the standard for patient age (<16mm at birth, <19mm at 12 months, and <21mm for adults (Table 1) as well as corneal diameter <10mm at birth) (1). TAL data considered normal is depicted in Table 2 (8). Microphthalmia may be associated with cataract, anterior segment malformation, corneal opacity, and posterior segment dysgenesis (3,9,10). It can be bilateral or unilateral. Additionally, its prognosis tends to be worse when accompanied by corneal opacity, aniridia, cataract, persistent fetal vasculature, and/or retinal dysplasia. Eyes with microphthalmia usually have a corneal diameter of <10mm; (Figure 1) however, when the corneal diameter is >10mm, it is more appropriately defined as nanophthalmos or posterior microphthalmia (11). Visual acuity depends on the extent of retinal involvement and presence of any other associated changes. These are usually eyes with high hyperopia, which may present staphyloma in the coloboma area (when present), with consequent high myopia.


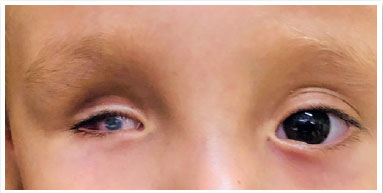
Anophthalmia is the complete absence of the eyeball in the presence of ocular adnexa (eyelids, conjunctiva, and tear ducts) (11,12). It is extremely rare and results from non-evagination of the optic vesicle. Furthermore, the optic nerve and tract are usually absent.
Another anomaly described in the literature is nanophthalmos, defined as axial length of the eyeball between 16 and 18mm without eye structure malformations. It features a shallow anterior chamber, high hyperopia (between 15 and 20D), and a normal-sized lens, which is large for this type of eyeball. Vessels can be tortuous and engorged, and there may be macular defects, chorioretinal pigment defects, and a “full” optic nerve. Corrected visual acuity may be normal during childhood, and prognosis depends on complications that may occur throughout life.
Coloboma is a segmental malformation resulting from failure of choroidal fissure closure; it can affect the iris, choroid, and ciliary body in the case of uveal coloboma, as well as the retina and optic nerve (Figures 2 and 3). One or more structures may present defects (13,14), which are located inferiorly in the inferonasal portion of the eyeball in the embryonic fissure closure pathway. Cyst formation can occur in the area of the defect, resulting in protrusion. Large colobomas may present with leukocoria, whereas small choroidal colobomas may resemble a whitish tumor, with a differential diagnosis of retinoblastoma in rare cases. Coloboma is the most common intraocular malformation in microphthalmia, and approximately 50% of the subjects with coloboma are affected bilaterally (8).
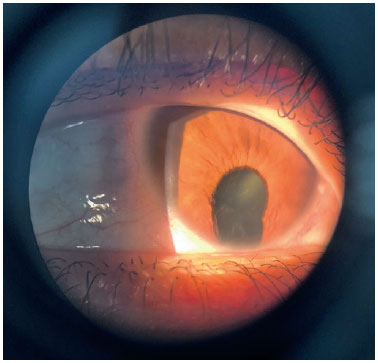
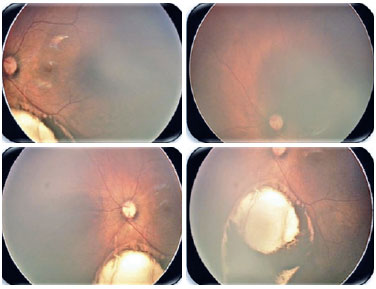
Diagnosis in the gestational period
Diagnosis during pregnancy can be performed using ultrasound (US). It is possible to detect anophthalmia/microphthalmia at the beginning of second trimester (3). Magnetic resonance imaging, when available, can also be used to supplement US (15).
Embryogenesis
During the 4 th week of pregnancy, the optic vesicle is formed through the evagination of neural tube epithelium (at the level of anterior diencephalon). The 5 th week features the budding of lens vesicle and invagination of optic vesicle, which morphs into a cup shape, resulting in the so-called “optic cup” stage. An invagination occurs in the ventral part of the optic vesicle, forming a fissure (through which the hyaloid artery and retinal axons pass) (13). Closure of the fissure begins at its midpoint, which is simultaneously proximal to the peduncle and distal to the optic cup edge; this process is completed within about seven weeks (13).
The occurrence of two events is important for enabling proper development process and avoiding coloboma: positioning of the edges and closure of the fissure. The gene expression pattern must be appropriate in time and space to cause fusion; however, a gene mutation may affect the cell-to-cell adhesion mechanism, preventing proper fusion even in the presence of anatomically well-positioned edges. In addition, the optic cup edges along the optic fissure should be juxtaposed with each other. A potential mutation may delay the growth of developing optic cup by not bringing the edges of the optic fissure close enough for their fusion (13).
In essence, when there is any failure in eye development resulting from an event that interrupts this process (between the 5 th and 7 th week of pregnancy), there may be unilateral or bilateral and symmetrical or asymmetrical malformations of the eyeball. When this failure occurs at a late stage, the only manifestation may be iris coloboma (13).
Classification
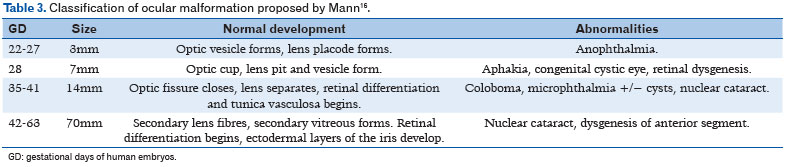
There are several classifications used in different studies. Over 50 years ago, Mann (16) proposed a classification for eye malformations according to presumed disruption of the development process and eye size in each gestational phase (Table 3) (6).
Another classification of the MAC spectrum is based on the phenotype determined by embryology and genetic etiology (Table 4). It divides conditions into anophthalmia in one or both eyes, microphthalmia without any optic fissure closure defects, microphthalmia without any optic fissure defect, coloboma in one or both eyes (with normal eye size), and unclassified microphthalmia. Iris coloboma was defined as an inferior defect of the iris or anterior stroma, whereas chorioretinal coloboma was defined as tissue absence in choroid and retina that may or may not extend to the optic disc. Microphthalmia may not be classified due to media opacity, which hinders the visualization of details.

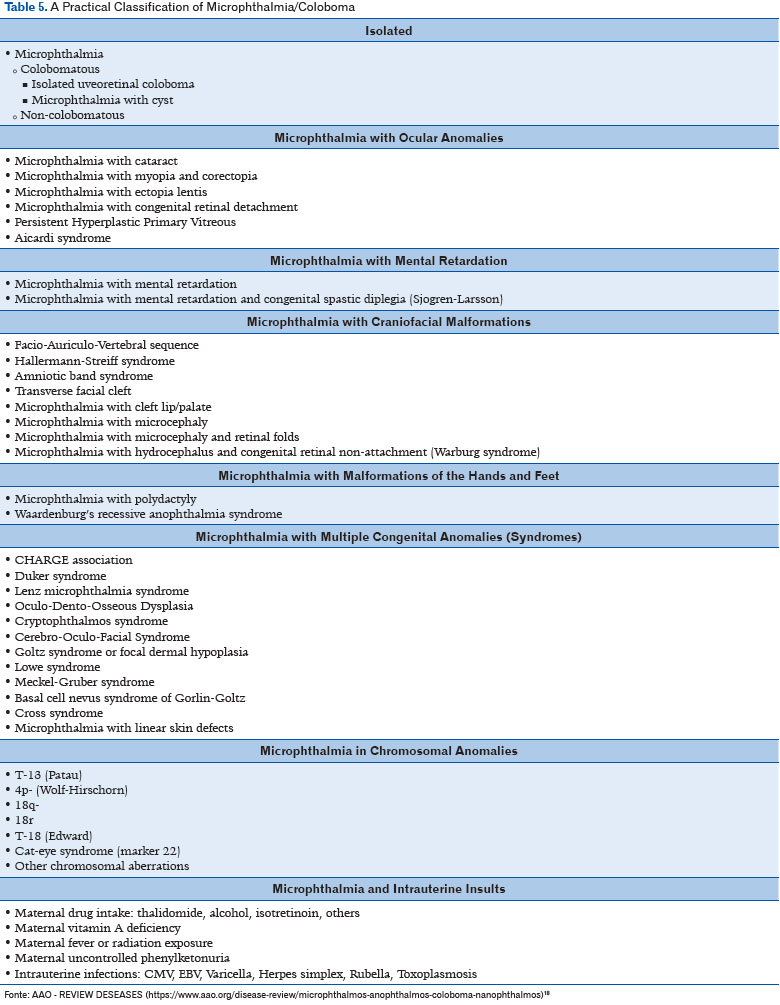
A third classification can be used more comprehensively, including other alterations associated with microphthalmia in addition to some risk factors and chromosomal changes (Table 5) (11).
Genetics
Eye development is an extraordinarily complex process and is controlled by a sequential and coordinated expression of genes. The genetic etiology of MAC spectrum is not well understood yet (5). Most MAC cases are sporadic, and genetic mutations are undetectable. However, clinical and experimental evidence suggests heterogeneous genetic involvement (18-21). The onset of eye development is controlled by a complex network of signaling molecules, transcription factors, structural proteins, and regulatory targets, which interact in specific pathways, forming differentiated tissues and cells in their due time of action (16,22,23). Therefore, superimposed phenotypes and clinical heterogeneity may exist as structural change can occur at any stage of eye development.
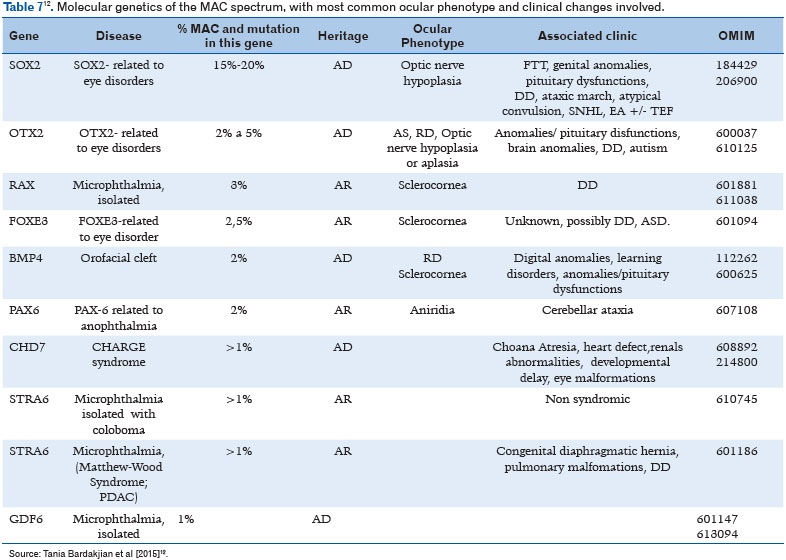
With the advent of gene and whole-exome sequencing, the underlying molecular basis have been discovered (11). More than 90 MAC-related genes have already been identified (24). Additionally, chromosomal abnormalities can be identified in an average of 25%-30% of subjects with MAC spectrum (Table 6), including trisomy of chromosome 9, 13, or 18 in addition to aneuploidy, triploidy, and microdeletion and microduplication syndromes (25). Table 7 describes the known genes involved, as well as inheritance, ocular phenotype, and the most frequent clinical associations (12).

GENETIC INHERITANCE
Sporadic cases are the most frequent; however, autosomal dominant, autosomal recessive, and X-linked recessive inheritance have also been described (11).
The genes most commonly involved in MAC are:
– Autosomal dominant pattern (AD): SOX2, OTX2, BMP4, PAX6, CHD7, TFAP2A11, and GDF6 (8);
– Autosomal recessive pattern (AR): RAX, FOXE3, STRA6, and SMOC1 (11);
– X-linked (XL) pattern: BCOR (11,8).
The genes least involved in MAC are:
– AD pattern: CRYBA4, HESX1, RARB, SHH, and TFAP2A (8);
– AR pattern: PXDN, SMOC1, VSX2, and TENM3 (8);
– XL pattern: HCCS, IKBKG, NAA10, NHS, and PORCN (8).
In cases of nanophthalmos and posterior microphthalmia, biallelic mutations are observed in PRSS56 or MFRP (11).
Isolated microphthalmia has been associated with the following gene loci (8) (Table 8):
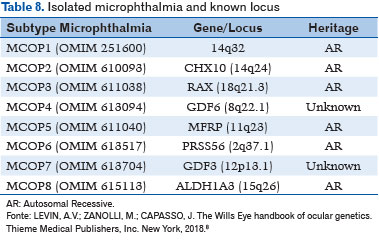
– AR pattern: CHX10, RAX, MFRP, PRSS56, and ALDH1A3;
– Unknown genetic inheritance: GDF6 and GDF3.
Severe bilateral cases are more likely to have an identifiable genetic cause (11,8), especially when environmental causes are not identified (11).
Risk factors
Some risk factors for MAC have been described, including drug use during pregnancy (such as thalidomide, alcohol, and isotretinoin), vitamin A deficiency, radiation exposure, uncontrolled maternal phenylketonuria, and maternal hypothyroidism (13). Other known causes are intrauterine infections (cytomegalovirus, Epstein-Barr virus, Herpes simplex virus, rubella, and toxoplasmosis) (11).
Diagnosis
Molecular genetic testing can identify the causative gene in about 20% of the subjects with MAC spectrum and around 80% of those with bilateral severe anophthalmia/microphthalmia (16,25,26). Chromosomal microarray analysis (CMA) or exome sequencing is indicated for cases with multiple malformations (11,27). Additionally, aneuploidy or chromosomal duplication, deletion, or rearrangement are investigated using CMA (12).
Another diagnostic method available in large centers is single gene testing, which includes sequence analysis and deletion or duplication analysis in addition to the analysis of genes that are more likely to mutate based on an individual’s clinical findings and/or family history (12).
Multigene panel testing for genes of interest may be requested. In some laboratories, clinicians indicate which multigene panel is most likely to identify the genetic cause of the disease at the most reasonable cost, whereas others have a custom panel designed by the laboratory and/or phenotype analysis including genes specified by the physician. Multigene panel methods may include sequence, duplication, and deletion analysis, or non-sequencing-based tests (12).
If single gene testing or multigene panel testing do not confirm the diagnosis in MAC spectrum, more comprehensive genomic assays may be considered, if available, including exome and mitochondrial sequencing (12).
Reevaluation in two years is recommended if no diagnosis is established after examinations and tests (12).
In cases of pregnancies with increased risk for MAC spectrum due to family history, including already having a child affected by these conditions, the test is based on genomic investigation of the affected relative (if a genetic alteration has been found). If a MAC spectrum anomaly is detected via US, additional diagnostic methods, such as CMA, SOX2 sequence analysis, deletion or duplication analysis, or a multigene panel, are recommended (12). Multigene panel assays allow a family-directed diagnosis, not only for the eye condition but also for potential systemic conditions (28). Families wherein pathogenic variants in a specific gene have already been identified, genetic testing of the embryo may be performed before implantation.
Eye complications and prognosis
Eye complications of microphthalmia/coloboma include angle-closure glaucoma, subretinal neovascularization, and, rarely, retinal detachment (11). Visual prognosis depends on the phenotype of each eye. In a study conducted by Hornby et al., isolated coloboma was reported to have the best prognosis, with good visual acuity in most cases. This was followed by coloboma with microcornea, which has a moderate prognosis; coloboma with microcornea and microphthalmia, which has a poor prognosis; and microphthalmia with cyst, which has the worst prognosis (27). It is noteworthy that, the volume of the eyeball triples between birth and adolescence under normal conditions (3,29).
Conduct
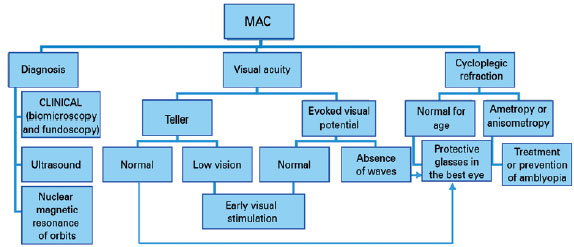
Treatment for MAC should be initiated within the first few weeks of life. The first step is diagnostic confirmation of the disease, and often additional examination, such as US imaging, is required.
The next step involves assessment of visual acuity and function. It is important to have cycloplegic refraction and prevent and/or treat amblyopia, instituting a conservative treatment. In unilateral cases, the “good eye” should be protected, with proper correction of its refractional changes (Diagram 1).
As microphthalmia and anophthalmia cause decreased orbital volume, most cases develop hemifacial asymmetry (since bone development depends on this volume). Reconstruction techniques are used to improve bone growth asymmetry and soft tissue hypoplasia (3,23).
In moderate microphthalmia, shapers (such as ocular prosthesis) have been used, which progressively increase in size to accompany bone development, favoring a more symmetrical development of the face. If possible, treatment should be initiated at around 6 months of age. In cases of severe microphthalmia and anophthalmia, expanders should be used in the first weeks of life to increase palpebral fissure, fundus of the conjunctival sac, and orbit (3,30). When the orbit develops properly, an ocular prosthesis can be used (Diagram 2).
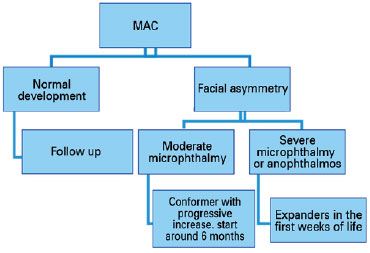
One or more surgical interventions may be required, and silicone balloon expanders, hydrophilic polymers, and dermis-fat grafts may be used as expanders.
In cases of microphthalmia with cyst, the approach depends on the degree of impairment, visual function, cyst size, and aesthetic condition of the patient. Aspiration of the cystic content or excision of the cyst can be performed with or without preservation of the eyeball.
Patients should be referred to a pediatric services for investigation of other associated systemic anomalies (31). A psychological assessment of patients and their families is important, as they can benefit from psychosocial support. Referral to a genetic specialist is also important for genetic counseling.
Given the variation in visual acuity, it is prudent not to predict visual acuity in babies based solely on appearance, as they can have surprisingly good vision. MAC impacts children’s lives significantly, similar to acute lymphoblastic leukemia and chronic systemic diseases (32).
DISCUSSION
Although various aspects about the etiology of MAC are yet to be elucidated and clarified, various studies are being conducted and knowledge on the same has been increasing exponentially. It is expected that novel treatment approaches will be possible at an early stage of eye development in the fetus, which may prevent the occurrence of genetic changes or failures during the vital period of eye formation.
REFERENCES
1. Reis LM, Semina EV. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today. 2015;105(2):96-113.
2. Ohuchi H, Sato K, Habuta M, Fujita H, Bando T. Congenital eye anomalies: More mosaic than thought? Congenit Anom (Kyoto). 2018 Jul 24. doi: 10.1111/cga.12304
3. Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007 Nov 26;2:47.
4. Hornby SJ, Gilbert CE, Rahi JK, Sil AK, Xiao Y, Dandona L, et al. Regional variation in blindness in children due to microphthalmos, anophthalmos and coloboma. Ophthalmic Epidemiol. 2000;7(2):127-38.
5. Ragge NK, Subak-Sharpe ID, Collin JRO. A practical guide to the management of anophthalmia and microphthalmia. Eye (Lond). 2007;21(10):1290-300.
6. Fitzpatrick DR, Van Heyningen V. Developmental eye disorders. Curr Opin Genet Dev. 2005;15(3):348-53.
7. Stoll C, Dott B, Alembik Y, Roth MP. Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol. 2012;94(3):147-52.
8. Levin AV, Zanolli M, Capasso J. The Wills Eye handbook of ocular genetics. Thieme Medical Publishers, Inc. New York, 2018.
9. Nishina S, Kurosaka D, Nishida Y, Kondo H, Kobayashi Y, Azuma N. Survey of microphthalmia in Japan. Jpn J Ophthalmol. 2012;56(3):198-202.
10. Shah SP, Taylor AE, Sowden JC, Ragge N, Russell-Eggitt I, Rahi JS, Gilbert CE, Surveillance of Eye Anomalies Special Interest Group. Anophthalmos, microphthalmos, and Coloboma in the United Kingdom: clinical features, results of investigations, and early management. Ophthalmology. 2012;119(2):362-8.
11. Khan AO, Traboulsi EI. AAO - Review Deseases. Microphthalmos, Anophthalmos, Coloboma, and Nanophthalmos (Includes CHARGE Association). A Compendium of Inherited Disorders and the Eye, Oxford University; 2016. https://www.aao.org/disease-review/microphthalmos-anophthalmos-coloboma-nanophthalmos
12. Bardakjian T, Weiss A, Schneider A. Microphthalmia/Anophthalmia/Coloboma Spectrum. 2004 Jan 29 [Updated 2015 Jul 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews ® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
13. Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-70.
14. Nakamura KM, Diehl NN, Mohney BG. Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol. 2011;129(1):69-74.
15. Brémond-Gignac D, Copin H, Elmaleh M, Milazzo S. Anomalies oculaires foetales: apport de l’imagerie anténatale en résonance magnétique [Fetal ocular anomalies: the advantages of prenatal magnetic resonance imaging]. J Fr Ophtalmol. 2010;33(5):350-4.
16. Mann I. The Developmental Basis of Eye Malformations. Philadelphia: JB Lippincott; 1953.
17. Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39(1):16-22
18. Mansoor N, Mansoor T, Ahmed M. Eye pathologies in neonates. Int J Ophthalmol. 2016;9(12):1832-8.
19. https://www.gov.uk/guidance/newborn-and-infant-physical-examination-screening-programme-overview Accessed Dec 26, 2015.
20. Nassetta L, Kimberlin D, Whitley R. Treatment of congenital cytomegalovirus infection: implications for future therapeutic strategies. J Antimicrob Chemother. 2009;63(5):862-7.
21. Malik AN, Hildebrand GD, Sekhri R, Russell-Eggitt IM. Bilateral macular scars following intrauterine herpes simplex virus type 2 infection. J AAPOS. 2008;12(3):305-6.
22. Kimberlin DW, Lin CY, Jacobs RF, Powell DA, Corey L, Gruber WC, Rathore M, Bradley JS, Diaz PS, Kumar M, Arvin AM, Gutierrez K, Shelton M, Weiner LB, Sleasman JW, de Sierra TM, Weller S, Soong SJ, Kiell J, Lakeman FD, Whitley RJ, National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics. 2001;108(2):230-8.
23. Tse DT, Pinchuk L, Davis S, Falcone SF, Lee W, Acosta AC, et al. Evaluation of an integrated orbital tissue expander in an anophthalmic feline model. Am J Ophthalmol. 2007;143:317-27.
24. Harding P, Moosajee M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J Dev Biol. 2019;7(3):16.
25. Chassaing N, Ragge N, Plaisancié J, Patat O, Geneviève D, Rivier F, et al. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Eur J Hum Genet. 2016;24(4):535-41.
26. Williamson KA, FitzPatrick DR. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet. 2014;57(8):369-80.
27. Raca G, Jackson CA, Kucinskas L, Warman B, Shieh JTC, Schneider A, et al. Array comparative genomic hybridization analysis in patients with anophthalmia, microphthalmia, and coloboma. Genet Med. 2011;13(5):437-42.
28. Patel A, Hayward JD, Tailor V, Nyanhete R, Ahlfors H, Gabriel C, et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalm.ology. 2019;126(6):888-907.
29. Skalicky SE, White AJR, Grigg JR, Martin F, Smith J, Jones M, et al. Microphthalmia, Anophthalmia, and Coloboma and Associated Ocular and Systemic Features: Understanding the Spectrum. JAMA Ophthalmol 2013;131(12):1517-24.
30. Clauser L, Sarti E, Dallera V, Galiè M. Integrated reconstructive strategies for treating the anophthalmic orbit. J Craniomaxillofac Surg. 2004;32:279-90.
31. Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol. 2010;94(4):487-93.
32. Dahlmann-Noor A, Tailor V, Abou-Rayyah Y, Adams G, Brookes J, Khaw SPT, et al. Functional vision and quality of life in children with microphthalmia/anophthalmia/coloboma-across-sectional study. J AAPOS. 2018;22(4):281-5.5.
AUTHOR’S INFORMATION
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
May 7, 2020.
Accepted on:
August 5, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket