

Ingrid de Oliveira Haubert Magalhães1; Priscila Hae Rim2; Marcelo Torigoe3
DOI: 10.17545/eOftalmo/2020.0017
RESUMO
As malformações do globo ocular são ocasionadas por defeito ou ruptura na fase de diferenciação e/ou desenvolvimento de tecidos e células especializadas. Neste artigo são abordadas principalmente três anomalias congênitas dos olhos, ou seja, microftalmia, anoftalmia e coloboma (MAC), que são malformações oculares estruturais congênitas que podem ser isoladas ou fazer parte de uma síndrome (associadas a anomalias craniofaciais, esqueléticas, genitourinárias, cerebrais, cardíacas, entre outras). Considerando o impacto importante das malformações do globo ocular na vida das crianças e de seus familiares, os autores realizaram uma revisão narrativa da literatura sobre o tema, incluindo suas principais classificações, os genes relacionados e as perspectivas atuais. Foram pesquisados na base de dados Bireme/Pubmed artigos sobre microftalmia, anoftalmia e coloboma. A partir dos estudos que vêm sendo realizados, espera-se que no futuro seja possível realizar novas abordagens terapêuticas durante uma fase precoce do desenvolvimento ocular no feto para evitar que alterações genéticas ou falhas ocorram nesse período importante da formação ocular.
Palavras-chave: Coloboma; Microftalmia; Anoftalmia; Anormalidades do olho.
ABSTRACT
Eyeball malformations are caused by defects or ruptures at the stage of differentiation and/or development of specialized tissues and cells. This article mainly addresses three congenital eye anomalies, i.e., microphthalmia, anophthalmia, and coloboma, which are congenital, structural eye malformations that can occur in isolation or as part of a syndrome (associated with craniofacial, skeletal, genitourinary, cerebral, or cardiac anomalies, among others). Considering the substantial impact of eyeball malformations on the lives of children and their families, we conducted a narrative literature review on the subject, including its main classifications, related genes, and current perspectives, using articles on microphthalmia, anophthalmia, and coloboma retrieved from the Bireme/PubMed database. Based on the studies that have been conducted, the possibility of using novel treatment approaches at an early stage of fetal eye development in the future is expected, preventing genetic alterations or failures during this vital period of eye formation.
Keywords: Coloboma; Microphthalmia; Anophthalmia; Eye abnormalities.
INTRODUÇÃO
A incidência mundial de MAC foi estimada em 6-13/100.000 nascidos vivos, sendo microftalmia de 2-17/100.000, anoftalmia 0,6-4,2/100.000 e coloboma 2-14/100.000 nascidos vivos (1).
As malformações do globo ocular são causadas por ruptura ou defeito no desenvolvimento e/ou diferenciação de tecidos e células especializados (2). A maioria cursa com baixa visão importante na infância (3,4), e causa importante impacto na vida das crianças e dos familiares.
Neste artigo, serão abordadas três das principais anomalias congênitas dos olhos, são elas: microftalmia, anoftalmia e coloboma (MAC). MAC são malformações oculares estruturais congênitas que podem ser isoladas ou fazer parte de uma síndrome (3,5,6). A incidência de outras anomalias associadas variou nos estudos entre um terço (1) a 90 por cento (7) dos casos. Quando o quadro é sindrômico pode apresentar anomalias craniofaciais, esqueléticas, renais, genitais, cardíacas, cerebrais e outras. Alterações neurológicas estão mais comumente associadas a anoftalmia, enquanto defeitos urológicos e genitais são mais prevalentes em pacientes com coloboma e microftalmia (1).
O objetivo desta revisão é conhecer melhor as classificações existentes e buscar informações sobre mapeamento genético, tratamento precoce e melhor acompanhamento dos pacientes.
MÉTODOS
Esta revisão narrativa da literatura foi realizada na base de dados Bireme/Pubmed, compreendendo artigos sobre microftalmia, anoftalmia e coloboma. A estratégia de busca incluiu as seguintes palavras-chaves: “microftalmia”, “anoftalmia”, “coloboma”, “malformações oculares congênitas”, “anomalia do globo ocular”, “MAC”, e várias combinações dessas palavras-chaves, entre os anos de 1950 a 2020. Foram encontrados 102 artigos e selecionados 33 deles, os quais compreendiam dados sobre MAC, incluindo definição, embriogênese, classificações, genes e síndromes associadas. Foram excluídos relatos de casos, estudos em fase experimental ou em animais e aqueles que não foram significativos do ponto de vista estatístico.
RESULTADOS
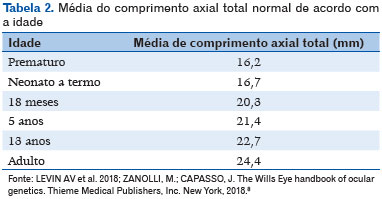
Microftalmia é um olho cujo comprimento axial total (CAT) é menor do que o padronizado para a idade do paciente (<16mm ao nascimento, <19mm aos 12 meses de vida, e <21mm no adulto - tabela 1 - e diâmetro corneal <10mm ao nascimento) (1). Os dados de CAT considerados normais encontram-se na tabela 2 (8); microftalmia pode estar associada a catarata, malformação do segmento anterior, opacidade da córnea e disgenesia do segmento posterior (3,9,10). Pode ser bilateral ou unilateral. Quando é acompanhada de opacidade corneal, aniridia, catarata, persistência da vasculatura fetal e/ou displasia retiniana, geralmente tem pior prognóstico. A córnea geralmente tem diâmetro menor que 10mm no olho com microftalmia; (Figura 1) e quando seu diâmetro é maior que 10mm, é mais apropriado definir o olho como nanoftalmo ou microftalmo posterior (11). A acuidade visual depende da extensão do acometimento retiniano e se existe alguma outra alteração associada. Usualmente são olhos com alta hipermetropia, porém podem apresentar um estafiloma na área do coloboma (quando existente), com consequente alta miopia.

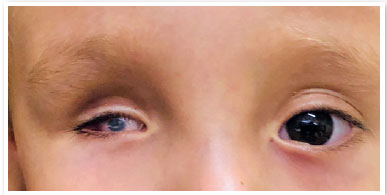
A anoftalmia se deve à ausência completa do globo ocular, na presença dos anexos oculares (pálpebras, conjuntiva e vias lacrimais) (11,12) É extremamente raro, e se dá pela não evaginação da vesícula óptica. Usualmente o nervo e trato ópticos estão ausentes.
Outra anomalia descrita é a nanoftalmia, definida como comprimento axial do globo ocular entre 16 e 18mm, sem malformações das estruturas oculares. A câmara anterior é rasa, tem alta hipermetropia (entre 15 e 20D), o cristalino tem geralmente tamanho normal, sendo grande para este tipo de globo. Os vasos podem ser tortuosos e ingurgitados, pode haver defeito macular, defeito pigmentar coriorretiniano, e disco “cheio”. A acuidade visual com correção pode ser normal na infância, e seu prognóstico depende das complicações que possam ocorrer ao longo da vida.
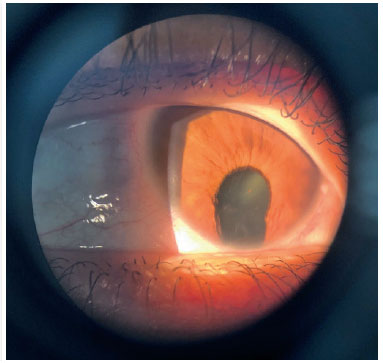
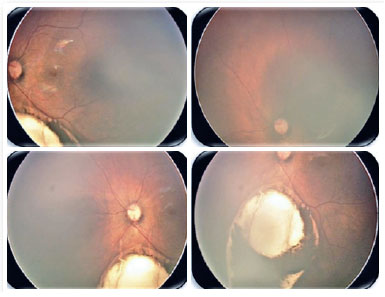
O coloboma é uma malformação segmentar devido falha no fechamento da fissura óptica, que pode afetar a íris, coróide e corpo ciliar - coloboma uveal, retina e nervo óptico (Figuras 2 e 3). Pode haver defeito em uma ou mais estruturas (13,14). Os defeitos são localizados inferiormente na porção ínfero-nasal do globo no trajeto de fechamento da fissura embrionária. Pode ser formado um cisto na área de defeito, acarretando uma protusão. Colobomas grandes podem apresentar leucocoria, e colobomas coroideanos pequenos podem parecer-se com tumor esbranquiçado, tendo diagnóstico diferencial o retinoblastoma, em raros casos. Coloboma é a malformação intraocular mais comum na microftalmia e aproximadamente 50% dos indivíduos com coloboma são bilateral (8).
Diagnóstico no período gestacional
O diagnóstico na gestação pode ser feito através de ultrassonografia (US). É possível detectar anoftalmia/microftalmia no início do segundo trimestre gestacional (3). A ressonância magnética, quando disponível, pode ser usada para suplementar a US (15).
Embriogênese
Durante a 4a semana de gestação há a formação da vesícula óptica, estrutura originada através da evaginação do epitélio do tubo neural (ao nível do diencéfalo anterior). Na 5a semana ocorre o brotamento da vesícula do cristalino e a invaginação da vesícula óptica, a qual assume um formato de copo, dando origem ao denominado estágio da “taça óptica”. Ocorre então uma invaginação na parte ventral da vesícula óptica, formando-se uma fissura (por onde passa a artéria hialóide e os axônios retinianos) (13). No ponto médio da fissura inicia-se seu fechamento, de forma simultânea proximal ao pedículo e distal à borda do cálice óptico, com conclusão deste processo por volta de 7 semanas (13).
Dois eventos devem ocorrer para o processo adequado do desenvolvimento evitando-se o coloboma: o posicionamento das bordas e o fechamento da fissura. O padrão de expressão gênica deve ser apropriado no tempo e espaço para provocar a fusão, ou uma mutação gênica pode afetar o mecanismo de adesão célula a célula, impedindo a fusão adequada mesmo na presença de bordas anatomicamente bem posicionadas. E as bordas do cálice óptico que margeiam a fenda óptica devem ficar justapostas entre si; pode ocorrer uma mutação que retarda o crescimento do cálice óptico em desenvolvimento não trazendo as bordas da fissura óptica próximas o suficiente para sua fusão (13).
Em suma, quando há alguma falha no desenvolvimento ocular, por qualquer evento que interrompa este processo, (entre a 5a e 7a semana de gestação) poderá haver a malformação unilateral ou bilateral, simétrico ou assimétrico do globo ocular. Quando esta falha se dá tardiamente, a única manifestação pode ser o coloboma isolado de íris (13).
Classificação
São diversas classificações utilizadas em vários estudos. Há mais de 50 anos, Mann (16) propôs uma classificação para as malformações oculares, de acordo com a interrupção presumida do processo de desenvolvimento, e tamanho do olho em cada fase gestacional (Tabela 3) (6).

Outra classificação dada ao espectro MAC, é baseada no fenótipo determinado pela embriologia e etiologia genética (Tabela 4). Foi dividida em anoftalmia em um ou ambos os olhos, microftalmia sem defeito no fechamento da fissura óptica, microftalmia sem defeito da fissura óptica, coloboma em um ou ambos os olhos (com tamanho de olho normal) e microftalmia não classificada. Coloboma de íris foi definido como um defeito inferior de íris ou do estroma anterior, coloboma coriorretiniano uma ausência tecidual de coróide e retina que pode ou não se extender para o disco óptico. A microftalmia pode não ser classificada devido opacidade de meios, não permitindo visualização de detalhes.

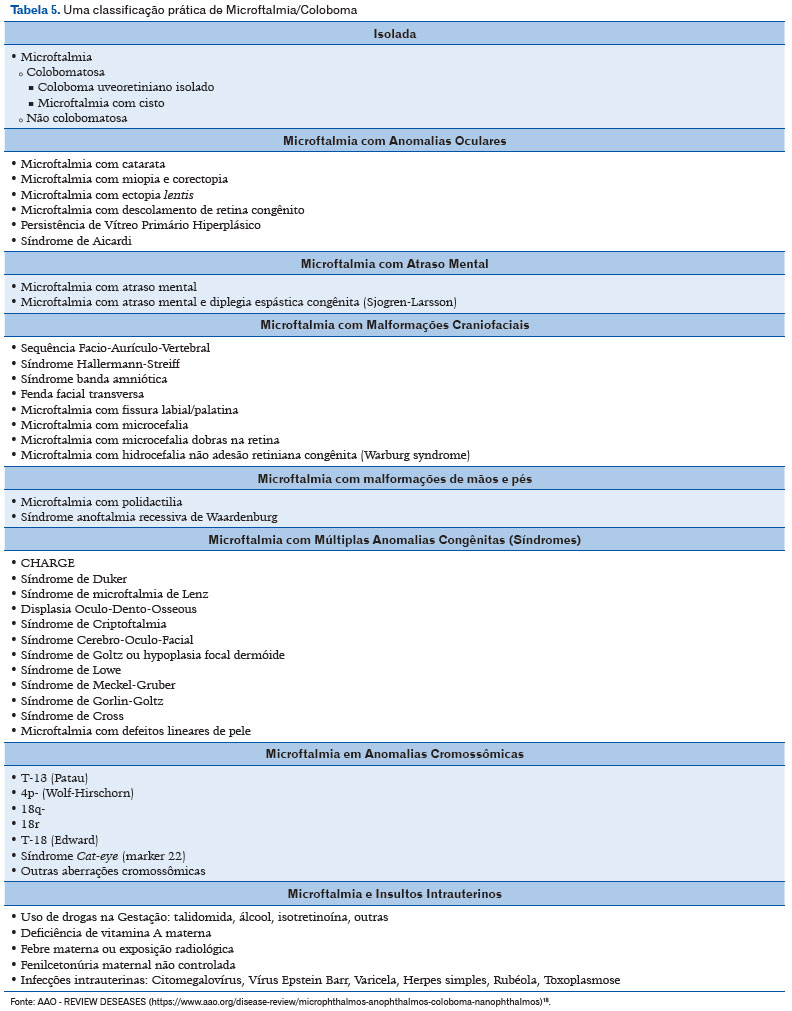
Uma terceira classificação pode ser utilizada, esta de forma mais abrangente, incluindo outras alterações associadas à microftalmia, assim como alguns fatores de risco e alterações cromossômicas (Tabela 5) (11).
Genética
O desenvolvimento do olho é altamente complexo. É determinado pela expressão sequencial e coordenada de genes. A etiologia genética do espectro MAC ainda não está muito bem compreendida (5). A maioria dos casos de MAC são esporádicos e as mutações genéticas não são detectáveis. No entanto, evidências clínicas e experimentais sugerem um envolvimento genético heterogêneo (18-21). O início do desenvolvimento ocular é controlado por uma complexa rede de moléculas de sinalização, fatores de transcrição, proteínas estruturais e alvos reguladores, que interagem em vias específicas, formando tecidos e células diferenciadas, no seu devido tempo de ação (16,22,23). Portanto, podem existir fenótipos sobrepostos e heterogeneidade clínica, uma vez que a alteração estrutural pode ocorrer em qualquer estágio do desenvolvimento ocular.
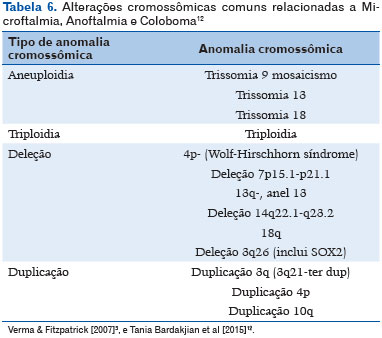
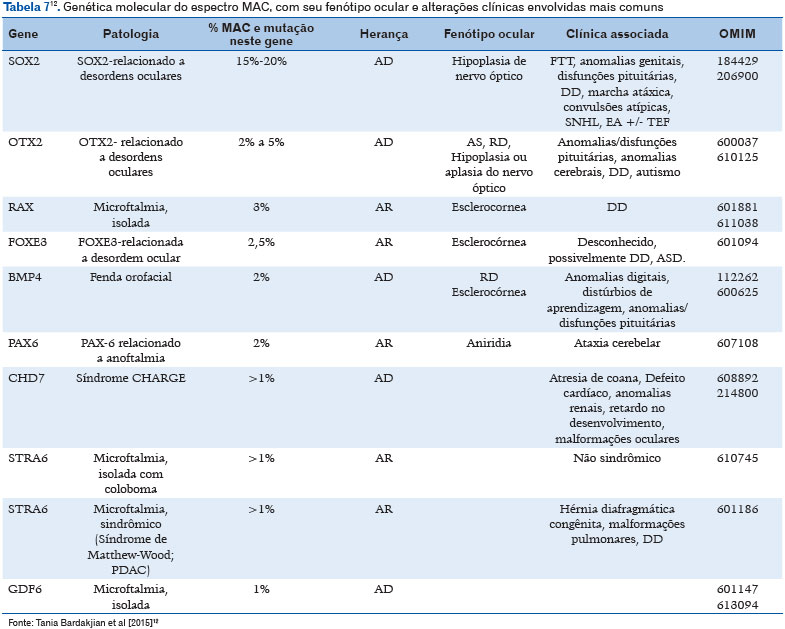
Com o advento do sequenciamento genético e sequenciamento completo do exoma a base molecular subjacente vem sendo descoberta (11). Mais de 90 genes relacionados com o MAC já foram identificados (24). As anormalidades cromossômicas podem ser identificadas em média de 25% a 30% dos indivíduos com o espectro MAC (Tabela 6), incluindo trissomia dos cromossomos 9, 13 ou 18, além de aneuploidia, triploidia e síndromes de microdeleção-duplicação (25). Na tabela 7 estão descritos os genes conhecidos envolvidos, assim como a herança, fenótipo ocular e associações clínicas mais frequentes (12).
Herança genética
Os casos esporádicos são os mais frequentes, entretanto estão descritas heranças autossômicas dominantes, autossômicas recessivas e recessivas ligadas ao X (11).
Os genes mais comumente envolvidos em MAC são:
– Padrão autossômico dominante (AD): SOX2, OTX2, BMP4, PAX6, CHD7, TFAP2A11; e GDF6 (8);
– Padrão autossômico recessivo (AR): RAX, FOXE3, STRA6, e SMOC1 (11);
– Ligado ao X (XL): BCOR (11,8).
Os genes menos envolvidos em MAC são:
– Padrão AD: CRYBA4, HESX1, RARB, SHH, TFAP2A (8)
– Padrão AR: PXDN, SMOC1, VSX2; TENM3 (8);
– XL: HCCS, IKBKG, NAA10, NHS, PORCN (8).
Nos casos de nanoftalmo e microftalmia posterior, observa-se mutações bialélicas em PRSS56 ou MFRP (11).
A microftalmia isolada tem os seguintes locus gênicos determinados (8) - Tabela 8 (8).
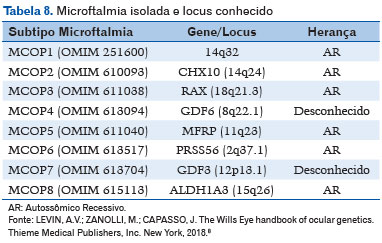
– Padrão AR: CHX10, RAX, MFRP, PRSS56 e ALDH1A3;
– Herança genética desconhecida: GDF6 e GDF3.
Aqueles casos severos bilaterais são mais propensos a ter uma causa genética identificável (11,8), quando não são causados por causas ambientais identificados (11).
Fatores de risco
São descritos alguns fatores de risco para a MAC: uso de drogas na gestação (como talidomida, álcool e isotretinoína), deficiência de vitamina A, exposição radiológica, fenilcetonúria materna descontrolada e hipotireoidismo materno (13). Outras causas conhecidas são as infecções intrauterinas (citomegalovírus, vírus Epstein-Barr, vírus Herpes Simples, rubéola e toxoplasmose) (11).
Diagnóstico
O teste genético molecular pode identificar o gene causador em torno de 20% dos indivíduos com espectro MAC e em torno de 80% com anoftalmia/microftalmia severa bilateral (16,25,26). A análise cromossômica por microarray (CMA) ou o sequenciamento do exoma é indicado nos casos com múltiplas malformações (11,27).
Através do CMA pesquisa-se a presença de aneuploidia ou duplicidade cromossômica, deleção ou rearranjo (12).
Outro teste disponível em grandes centros é o teste de gene único, este inclui análise de seqüência, bem como análise de deleção ou duplicação, e os genes que têm maior probabilidade de sofrer mutação, com base nos achados clínicos e/ou história familiar do indivíduo (12).
O painel multigênico é solicitado com a inclusão dos genes de interesse. Em alguns laboratórios os clínicos indicam qual painel multigênico tem maior probabilidade de identificar a causa genética da doença ao custo mais razoável; enquanto em outros laboratórios existe um painel personalizado projetado pelo laboratório e/ou a análise do fenótipo que inclua os genes especificados pelo médico. Os métodos no painel multigênico podem incluir análise de sequência, duplicação, deleção ou outros testes, que não baseados em sequenciamento (12).
Caso o teste de gene único ou o painel multigênico não confirmar um diagnóstico no espectro MAC, podem ser considerados ensaios genômicos mais abrangentes, se disponíveis, incluindo: sequenciamento do exoma e sequenciamento mitocondrial (12).
A reavaliação em dois anos é recomendada caso nenhum diagnóstico seja estabelecido após os exames e os testes (12).
Nos casos de gestações com risco aumentado para o espectro MAC devido a existência de uma criança anteriormente afetada e/ou se tiver histórico familiar, o teste baseia-se na investigação genômica presente no parente afetado (caso tenha sido encontrada a alteração genética). Quando uma anomalia do espectro MAC é detectada no US, é recomendado o diagnóstico adicional como o CMA, análise de sequência SOX2, análise deleção ou duplicação ou painel multigênico (12). Os ensaios com painéis multigênicos permitem um diagnóstico direcionado às famílias, não apenas para a condição ocular, mas também para condições sistêmicas potenciais (28).
Naquelas famílias que variantes patogênicas em um gene específico já foram identificadas pode ser realizado estudo genético do embrião antes da implantação do mesmo.
Complicações oculares e prognóstico
Complicações oculares de microftalmia/coloboma incluem glaucoma de ângulo fechado, neovascularização subretiniana e, raramente, descolamento de retina (11). O prognóstico visual depende do fenótipo de cada olho. Em um estudo realizado por Hornby et al, foi observado que o coloboma isolado tem o melhor prognóstico, com boa acuidade visual em sua maioria dos casos, seguido de coloboma com microcórnea, que tem prognóstico moderado; Já o coloboma com microcórnea e microftalmo tem prognóstico ruim, enquanto o microftalmo com cisto tem o pior prognóstico (27). É importante notar que em condições normais o volume do globo ocular triplica entre o nascimento e a adolescência (3,29).
Conduta
O tratamento da MAC deve ser iniciado nas primeiras semanas de vida. O primeiro passo é a confirmação diagnóstica da doença, e muitas vezes é necessário exame complementar, como a ultrassonografia.
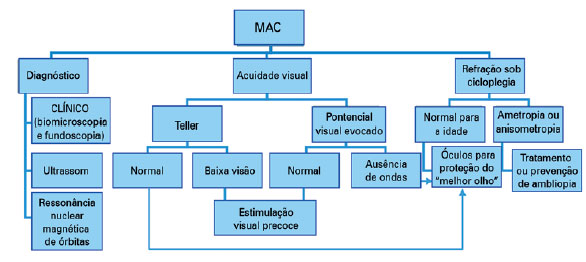
O estudo da acuidade e função visual é o próximo passo. É importante refratar os olhos sob cicloplegia e prevenir/tratar a ambliopia, instituindo o tratamento conservador. Nos casos unilaterais deve-se proteger o “olho bom” e corrigir apropriadamente suas alterações refracionais (Diagrama 1).
Como a microftalmia e anoftalmia cursam com diminuição do volume orbitário, evolui na maioria dos casos com assimetria hemifacial (pois o desenvolvimento ósseo depende deste volume). Técnicas de reconstrução são utilizadas para uma melhora da assimetria de crescimento ósseo e hipoplasia de tecidos moles (3,23).
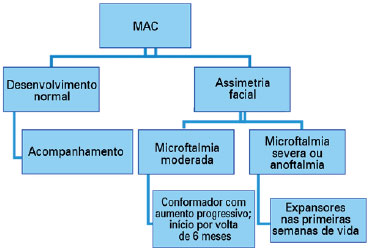
Na microftalmia moderada tem-se utilizado um conformador (como uma prótese ocular), com aumento progressivo de seu tamanho para acompanhar o desenvolvimento ósseo, favorecendo um desenvolvimento mais simétrico da face, e, se possível, o tratamento deve ter início por volta de 6 meses. Já nos casos de microftalmia severa e anoftalmia devem ser utilizados nas primeiras semanas de vida expansões para aumentar a fissura palpebral, fundo de saco conjuntival e a órbita (3,30). Quando a órbita se desenvolve adequadamente pode-se utilizar a prótese ocular (Diagrama 2).
Pode ser necessário uma ou mais intervenções cirúrgicas, e serem utilizados como expansores o balão de silicone, polímeros hidrofílicos, e enxertos de tecido dermo-gorduroso.
Nos casos de microftalmia com cisto a abordagem depende do grau de acometimento, função visual, tamanho do cisto, e estética do paciente. Há opção de ser aspirado o conteúdo cístico ou realizar a excisão cística com ou sem a preservação do globo.
Deve-se referenciar ao serviço de pediatria para investigação de outras anomalias sistêmicas associadas (31). Importante ainda avaliar a parte psicológica tanto do paciente quanto de seus familiares, pois estes podem se beneficiar de apoio psicossocial. Encaminhar ao especialista em genética também é importante para aconselhamento genético.
Tendo em vista que a acuidade visual é variada, é prudente não predizer sobre a acuidade visual nos bebês baseada somente na aparência, pois estes podem surpreender com boa visão. MAC tem impactos importantes na vida das crianças, sendo similar àqueles descritos por aquelas com leucemia linfoblástica aguda e com doenças sistêmicas crônicas (32).
DISCUSSÃO
Há muito a ser esclarecido sobre a etiologia do MAC, no entanto muitos estudos estão sendo realizados e o conhecimento vem aumentando exponencialmente. No futuro, espera-se que seja possível realizar novas abordagens terapêuticas durante uma fase precoce do desenvolvimento ocular no feto para evitar que alterações genéticas ou falhas ocorram nesse período importante da formação ocular.
REFERÊNCIAS
1. Reis LM, Semina EV. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today. 2015;105(2):96-113.
2. Ohuchi H, Sato K, Habuta M, Fujita H, Bando T. Congenital eye anomalies: More mosaic than thought? Congenit Anom (Kyoto). 2018 Jul 24. doi: 10.1111/cga.12304
3. Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007 Nov 26;2:47.
4. Hornby SJ, Gilbert CE, Rahi JK, Sil AK, Xiao Y, Dandona L, et al. Regional variation in blindness in children due to microphthalmos, anophthalmos and coloboma. Ophthalmic Epidemiol. 2000;7(2):127-38.
5. Ragge NK, Subak-Sharpe ID, Collin JRO. A practical guide to the management of anophthalmia and microphthalmia. Eye (Lond). 2007;21(10):1290-300.
6. Fitzpatrick DR, Van Heyningen V. Developmental eye disorders. Curr Opin Genet Dev. 2005;15(3):348-53.
7. Stoll C, Dott B, Alembik Y, Roth MP. Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol. 2012;94(3):147-52.
8. Levin AV, Zanolli M, Capasso J. The Wills Eye handbook of ocular genetics. Thieme Medical Publishers, Inc. New York, 2018.
9. Nishina S, Kurosaka D, Nishida Y, Kondo H, Kobayashi Y, Azuma N. Survey of microphthalmia in Japan. Jpn J Ophthalmol. 2012;56(3):198-202.
10. Shah SP, Taylor AE, Sowden JC, Ragge N, Russell-Eggitt I, Rahi JS, Gilbert CE, Surveillance of Eye Anomalies Special Interest Group. Anophthalmos, microphthalmos, and Coloboma in the United Kingdom: clinical features, results of investigations, and early management. Ophthalmology. 2012;119(2):362-8.
11. Khan AO, Traboulsi EI. AAO - Review Deseases. Microphthalmos, Anophthalmos, Coloboma, and Nanophthalmos (Includes CHARGE Association). A Compendium of Inherited Disorders and the Eye, Oxford University; 2016. https://www.aao.org/disease-review/microphthalmos-anophthalmos-coloboma-nanophthalmos
12. Bardakjian T, Weiss A, Schneider A. Microphthalmia/Anophthalmia/Coloboma Spectrum. 2004 Jan 29 [Updated 2015 Jul 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews ® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
13. Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-70.
14. Nakamura KM, Diehl NN, Mohney BG. Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol. 2011;129(1):69-74.
15. Brémond-Gignac D, Copin H, Elmaleh M, Milazzo S. Anomalies oculaires foetales: apport de l’imagerie anténatale en résonance magnétique [Fetal ocular anomalies: the advantages of prenatal magnetic resonance imaging]. J Fr Ophtalmol. 2010;33(5):350-4.
16. Mann I. The Developmental Basis of Eye Malformations. Philadelphia: JB Lippincott; 1953.
17. Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39(1):16-22
18. Mansoor N, Mansoor T, Ahmed M. Eye pathologies in neonates. Int J Ophthalmol. 2016;9(12):1832-8.
19. https://www.gov.uk/guidance/newborn-and-infant-physical-examination-screening-programme-overview Accessed Dec 26, 2015 .
20. Nassetta L, Kimberlin D, Whitley R. Treatment of congenital cytomegalovirus infection: implications for future therapeutic strategies. J Antimicrob Chemother. 2009;63(5):862-7.
21. Malik AN, Hildebrand GD, Sekhri R, Russell-Eggitt IM. Bilateral macular scars following intrauterine herpes simplex virus type 2 infection. J AAPOS. 2008;12(3):305-6.
22. Kimberlin DW, Lin CY, Jacobs RF, Powell DA, Corey L, Gruber WC, Rathore M, Bradley JS, Diaz PS, Kumar M, Arvin AM, Gutierrez K, Shelton M, Weiner LB, Sleasman JW, de Sierra TM, Weller S, Soong SJ, Kiell J, Lakeman FD, Whitley RJ, National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics. 2001;108(2):230-8.
23. Tse DT, Pinchuk L, Davis S, Falcone SF, Lee W, Acosta AC, et al. Evaluation of an integrated orbital tissue expander in an anophthalmic feline model. Am J Ophthalmol. 2007;143:317-27.
24. Harding P, Moosajee M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J Dev Biol. 2019;7(3):16.
25. Chassaing N, Ragge N, Plaisancié J, Patat O, Geneviève D, Rivier F, et al. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Eur J Hum Genet. 2016;24(4):535-41.
26. Williamson KA, FitzPatrick DR. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet. 2014;57(8):369-80.
27. Raca G, Jackson CA, Kucinskas L, Warman B, Shieh JTC, Schneider A, et al. Array comparative genomic hybridization analysis in patients with anophthalmia, microphthalmia, and coloboma. Genet Med. 2011;13(5):437-42.
28. Patel A, Hayward JD, Tailor V, Nyanhete R, Ahlfors H, Gabriel C, et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology. 2019;126(6):888-907.
29. Skalicky SE, White AJR, Grigg JR, Martin F, Smith J, Jones M, et al. Microphthalmia, Anophthalmia, and Coloboma and Associated Ocular and Systemic Features: Understanding the Spectrum. JAMA Ophthalmol. 2013;131(12):1517-24.
30. Clauser L, Sarti E, Dallera V, Galiè M. Integrated reconstructive strategies for treating the anophthalmic orbit. J Craniomaxillofac Surg. 2004;32:279-90.
31. Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol. 2010;94(4):487-93.
32. Dahlmann-Noor A, Tailor V, Abou-Rayyah Y, Adams G, Brookes J, Khaw SPT, et al. Functional vision and quality of life in children with microphthalmia/anophthalmia/coloboma-a cross-sectional study. J AAPOS. 2018;22(4):281-5.5.
INFORMAÇÃO DOS AUTORES
Financiamento: Declaram não haver.
Conflitos de Interesse: Declaram não haver.
Recebido em:
7 de Maio de 2020.
Aceito em:
5 de Agosto de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket