

Simone Cristine Hermes Hoff1; Aline Christina Fioravanti Lui2; Eliza Antonieta Rosana Negrão Grangeiro3
DOI: 10.17545/e-oftalmo.cbo/2016.63
ABSTRACT
Acute posterior multifocal pigment epitheliopathy (apmppe) is classified as white dot syndrome, It is rare and self-limited and involves low visual acuity and sudden multiple placoid lesions, which are bilateral and asymmetrical, The case of a 50 year-old patient with sudden visual loss was reported (visual acuity in both eyes: counting the number of fingers at 1 meter), with alterations in the fundus and fluorescein angiography results similar to that observed in vogt-koyanagi-harada syndrome, with multiple areas of bilateral, asymmetric retinal detachment involving the macular area and periphery, Differential diagnosis was made based on the absence of panuveitis and/or extraocular findings, and good final visual acuity (20/30 in both eyes) was observed, even without treatment.
Keywords: Choroiditis; Uveomeningoencephalitic Syndrome; Fluorescein angiography; Retinal detachment
RESUMO
A epiteliopatia pigmentar placoide multifocal posterior aguda (EPPMPA) é uma doença incluída na síndrome dos pontos brancos; é rara e autolimitada, cursa com baixa de acuidade visual (BAV) súbita e múltiplas lesões placoides, bilateral e assimétrica. Descreve-se aqui um caso de uma paciente de 50 anos, com BAV súbita (AV AO: CD a 1 m), com achados fundoscópicos e angiofluoresceinográficos muito semelhantes à Síndrome de Vogt-Koyanagi-Harada, com múltiplas áreas de descolamento de retina, bilaterais, assimétricas, envolvendo a área macular e a periferia. A diferenciação diagnóstica foi feita baseada na ausência de pan-uveíte e/ou achados extraoculares, e boa AV final (20/30 AO), mesmo na ausência de tratamento.
Palavras-chave: Coroidite; Síndrome Uveomeningoencefálica; Angiofluoresceinografia; Descolamento Retiniano
INTRODUCTION
The white dot syndrome comprises a heterogeneous group of syndromes characterized by multiple posterior pole lesions with inflammation of the choroid, retinal pigment epithelium, and retina. 1
One of the diseases included in this syndrome is acute posterior multifocal placoid pigment epitheliopathy (APMPPE), which is a rare and self-limiting disease, typically affecting young adults (20-50 years of age).2,3 This epitheliopathy was originally described by Gass (1968) as a pathology mainly affecting RPE, of little known etiology and pathogenesis, abrupt onset, and multiple bilateral, asymmetric placoid lesions, resulting in sudden low visual acuity (LVA).1 Traditional treatment consists of 3-6 weeks of oral corticosteroid therapy, evolving with good final visual prognosis.1
CASE REPORT
A 50-year-old Caucasian female patient, born and living in São Paulo, was admitted to the hospital emergency room complaining of sudden LVA in both eyes 2 days ago. She did not suffer from systemic arterial hypertension, diabetes, headache, tinnitus, or history of ophthalmological pathology.
On visual test, she presented with VA in both eyes for counting fingers at 1 m, normal biomicroscopy, and IOP.
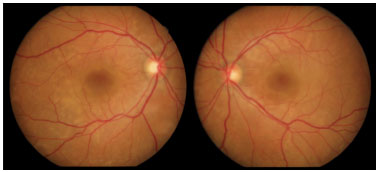
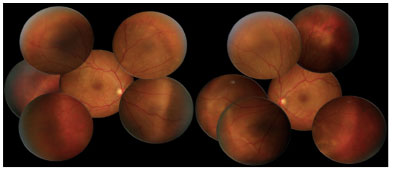
Retinography on admission revealed multiple areas of bilateral, asymmetric retinal detachment affecting the macular area and periphery of the retina (Figures 1 and 2).
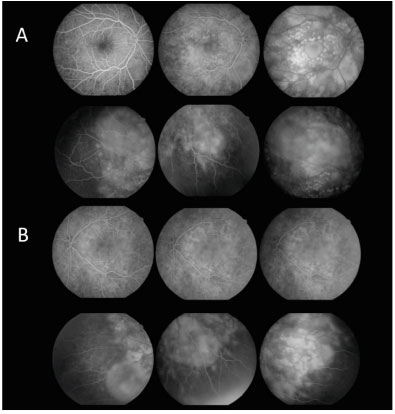
Fluorescein angiography (FA) revealed multiple areas of bilateral, asymmetric late hyperfluorescence involving the macular area and periphery of the retina, representing serous RD and RPE detachment. Late phase with multiple areas of hypofluorescence was observed (Figure 3).
A hemogram, ESR, RF, PPD, serological tests for toxoplasmosis, VDRL and FTA-ABS, chest X-ray, and chest and cranial CT were requested, and all results were within the normal ranges. Oral prednisone (1 mg/Kg/day) was prescribed.
After 12 days, the patient returned with VA in both eyes of c/c 20/30, without having taken the prescribed medication. Retinography was performed at 12 and 30 days of evolution showing significant improvement in the condition, followed by spontaneous resolution (Figure 4). FA at 30 days of evolution showed multiple punctiform window defect areas of hypofluorescence corresponding to the perimacular scar tissue with resolution of the multiple retinal detachments in the macular area and periphery of the retina (Figure 5).
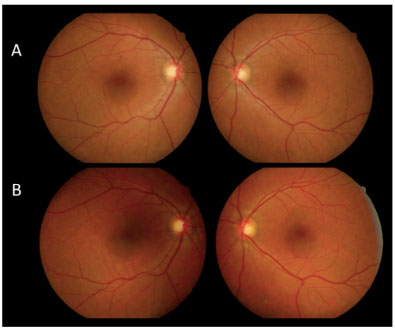
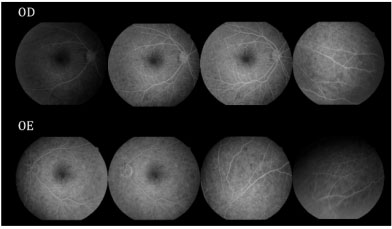
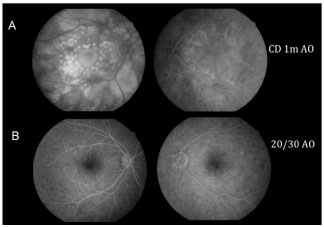
Figure 6 compares FA of the macular area of both eyes at the initial stage of the condition with VA in both eyes for counting fingers at 1 m and after 30 days without treatment with good final VA (20/30 in both eyes).
DISCUSSION
APMPPE is a rare disease that presents as multiple bilateral, asymmetric placoid lesions causing sudden LVA.3 During FA, it presents as early hypofluorescence or hyperfluorescence, with hyperfluorescence that increases in intensity and size because of extravasation of contrast in the late phase.4,5
The Vogt-Koyanagi-Harada Syndrome (VKHS) has a probable autoimmune etiology that manifests as multiple bilateral, asymmetric retinal detachments, resulting in serious vision loss if not treated properly.6 The complete syndrome presents as neurological, hearing, and skin changes in addition to panuveitis, with no previous history of eye disease, surgery, or trauma.7 During FA, areas of hyperfluorescence in the early phase and extravasation in the late phase can be observed, which result from a buildup of subretinal fluid.6
When APMPPE involves areas of serous retinal detachments, diagnostic differentiation is difficult because of the similarity in FA findings between VKHS and APMPPE. The patient described herein had a confirmed diagnosis of APMPPE because there was no panuveitis or other extraocular changes and because her evolution was favorable with good final visual acuity (20/30 in both eyes) and improvement in the funduscopic and FA changes, even though she did not receive any treatment.
REFERENCES
1. Gross NE, Yannuzzi LA, Freund KB, Spaide RF, Amato GP, Sigal R. Multiple evanescent white dot syndrome. Arch Ophthalmol. 2006;124(4):493-500. http://dx.doi.Org/10.1001/archopht.124.4.493
2. Durrani A, Rahimy E, Dunn JP. Bilateral acute-onset vision loss in a previously healthy man. JAMA Ophthalmol. 2015;133:957-8. http://dx.doi.org/10.1001/jamaophthalmol.2015.1139
3. Mrejen S, Gallego-Pinazo R, Wald KJ, Freund KB. Acute posterior multifocal placoid pigment epitheliopathy as a choroidopathy: what we learned from adaptive optics imaging. JAMA Ophthalmol. 2013;131:1363-4. http://dx.doi.org/10.1001/jamaophthalmol.2013.4196
4. Fine HF, Spaide RF, Ryan Jr EH, Matsumoto Y, Yannuzzi LA. Acute zonal occult outer retinopathy in patients with multiple evanescent white dot syndrome. Arch Ophthalmol. 2009;127:66-70. http://dx.doi.org/10.1001/archophthalmol.2008.530
5. Yeh S, Forooghian F, Wong WT, Faia LJ, Cukras C, Lew JC, et al. Fundus autoflourescence imaging of the white dot syndromes. Arch Ophthalmol. 2010;128:46-56. http://dx.doi.org/10.1001/archophthalmol.2009.368
6. Mota LAA, Santos AB. Síndrome de Vogt-Koyanagi-Harada e o seu acometimento multissistêmico. Rev Assoc Med Bras. 2010; 56(5):590-5. http://dx.doi.org/10.1590/S0104-42302010000500023
7. Lucena DR, Paula JS, Silva GCM, Rodrigues MLV. Síndrome de Vogt-Koyanagi-Harada incompleta associada a HLA DRB1*01 em criança de quatro nos de idade: relato de caso. Arq Bras Oftalmol. 2007;70(2):340-2. http://dx.doi.org/10.1590/S0004-27492007000200027



Funding source: None
Conflicts of interest: None
Received on:
October 24, 2016.
Accepted on:
November 24, 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket