

Francisco Victor Carvalho Barroso1; Ana Laura Eloia Limão2; Rian Vilar Lima3; Samuel Montenegro4; Manoela Gondim5; João Crispim Moraes Lima Ribeiro2
DOI: 10.17545/eOftalmo/2024.0014
ABSTRACT
Through this literature review, we aimed to determine the ocular alterations in patients with Dandy-Walker syndrome. Embase, Medline, Lilacs, and Scielo databases were searched using the following key words "Eye" and "Dandy-Walker" without any limits. The search yielded 1110 articles, which was reduced to 109 articles after non-relevant articles were excluded. The full text of 54 articles were read, and 22 studies were finally evaluated. Approximately 57% and 42% of all the patients in our sample exhibited anterior segment and posterior segment alterations, respectively. However, high impact studies are required to better describe the associations between ocular manifestations and Dandy-Walker syndrome.
Keywords: Dandy-Walker syndrome; Mega cisterna magna; Eye abnormalities; Ophthalmologic; Ophthalmology.
RESUMO
O objetivo desta revisão de literatura é estudar as alterações oculares em pacientes com Síndrome de Dandy-Walker. Uma revisão da literatura foi feita usando as bases de dados Embase, Medline, Lilacs e Scielo, pesquisando artigos usando as seguintes palavras gerais "Eye" e "Dandy-Walker" como termos de busca, sem limitar onde esses termos foram encontrados em diferentes seções do artigo. A busca encontrou 109 artigos e após critérios de exclusão 1.110 artigos. Foram selecionados 54 registros para leitura na íntegra e finalmente incluídos 22 estudos para descrever as associações. 57% de todos os pacientes da nossa amostra apresentaram alteração do segmento anterior e 42% alteração do segmento posterior. Estudos de alto impacto são necessários para melhor descrever as associações mencionadas.
Palavras-chave: Síndrome de Dandy-Walker; Mega cisterna magna; Anomalias oculares; Oftalmológica; Oftalmologia.
INTRODUCTION
Dandy-Walker syndrome (DWS) is a rare posterior fossa malformation that develops in 1 in 25 000-30 000 live births1,2. The most frequent malformations of the posterior fossa are mega cisterna magna (31.8%) and Dandy-Walker malformation (27%)3. DWS typically occurs sporadically and was first described in 1954. Considering the development of cerebellar vermis as the standard, DWS can be categorized as follows: Dandy-Walker malformation, Dandy-Walker variant, and simple posterior fossa cistern widening. However, individual physical abnormalities should be known. Although several patients remain clinically asymptomatic for years, others may present with a variety of comorbidities leading to an earlier diagnosis4. DWS is associated with chromosome 3 deletions, which can cause cystic dilation of the fourth ventricle during the 4th month of fetal life5. Thus, the patients can present with ocular features such as hypertelorism, epicanthus, glaucoma, and anophthalmia, which are related to chromosome 3 mutations6. Other gene mutations, such as those of ZIC, ZIC4, FOXC1, FGF17, LAMC1, and NID17-13, have also been reported in association with DWS. Furthermore, the deletion of different chromosomes, including 3, 6, and 13, have been reported in DWS. The deletion interval on chromosome 13 is the 13q32.2-33.2 region, which codes for the ZIC2 and ZIC5 genes13. G9a, also known as euchromatin histone methyltransferase, is a protein lysine methyltransferase that induces methylation of several proteins, including histones. G9a has also been associated with DWS. Nonetheless, none of these factors are related to eye embryogenesis13.
The classic anatomic hallmarks of DWS are hypoplasia of the cerebellar vermis, anteroposterior enlargement of the posterior fossa, and cystic dilation of the fourth ventricle. The DWS variant is defined as the absence of tentorium dislocation, torcular-lambdoid inversion, or vermian rotation. The treatment of DWS is generally focused on alleviating hydrocephalus and posterior fossa symptoms, often including surgical interventions such as ventriculoperitoneal and cystoperitoneal shunting. However, a systematic literature review on the ocular manifestations of DWS is lacking. Therefore, herein, we aimed to systematically review all the reported cases in the literature.
METHODS
Literature search
This study followed an exploratory-descriptive and qualitative approach and used the methods described in the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) 2020 protocol (Figure 1). The MEDLINE (via PubMed), Lilacs and Scielo (via BVS), Embase, and Web of Science databases were last searched on March 10, 2023. Each platform was searched using the appropriate descriptors, without considering a specific language or time frame. The following descriptors (MeSH) were searched using the boolean operators AND and OR: "Dandy-Walker Syndrome," "DWS," "mega cisterna magna," "eye," "eye abnormalities," "ophthalmologic," "ophthalmology," and "ophthalmic." The references of all the included studies were also reviewed for additional relevant publications.
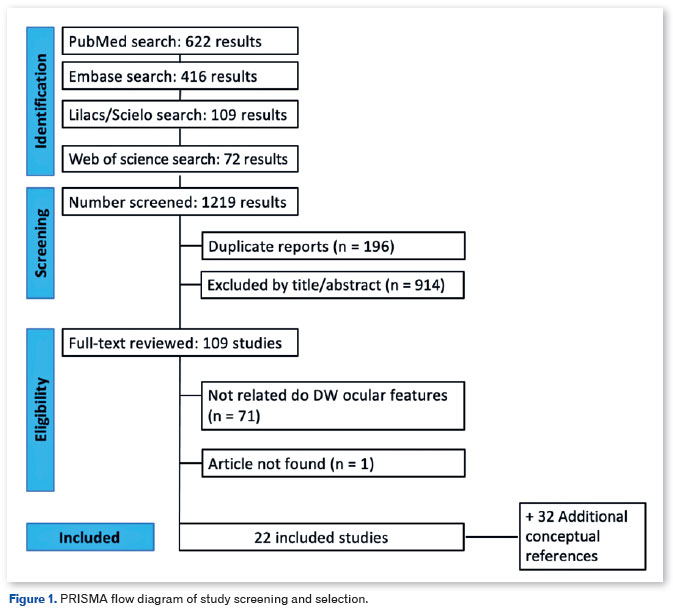
Eligibility criteria
Initially, 1110 articles were identified and independently screened by four investigators. Possible disagreements were resolved by a fifth investigator. After excluding duplicate articles, 1023 studies remained. The titles and abstracts of the 1023 studies were evaluated according to the PECOT model for inclusion or exclusion. The inclusion criteria were as follows: (a) participants: patients with DWS; (b) exposure: development of ocular abnormalities; (c) control: none; (d) outcome: type of abnormality, segment of the abnormality, and associated genes; and (e) study design: observational studies. The exclusion criteria were as follows: (a) studies that did not report ophthalmic abnormalities or any ophthalmological assessment; (b) population overlap between publications; and (c) unavailability of full texts or complete data after contacting the original authors. To ensure that the review was inclusive of all relevant findings, studies that reported patients with Dandy-Walker anomaly were also included even if the diagnosis was not radiologically confirmed. Although the quality of the studies was not an exclusion criterion, it was duly acknowledged in the results.
Data extraction
Two authors independently extracted the data from all the eligible studies. To ensure the integrity and accuracy of the information, disagreements were discussed and resolved by consensus with a third reviewer. We extracted the following data from the eligible studies: first author's last name, year of publication and country of study, data sources, duration of follow-up, age range, number of patients, mean or median age, genetic abnormalities, and eye abnormalities in the anterior and posterior segments.
Data analysis
The variables are expressed qualitatively. For the continuous variables, a purely descriptive approach was adopted (i.e., data are expressed as unweighted means whenever possible). No further statistical analyses were performed.
Ethical aspects
Because the articles analyzed in this study are publicly available in scientific databases and portals, the need for ethics approval was waived.
RESULTS
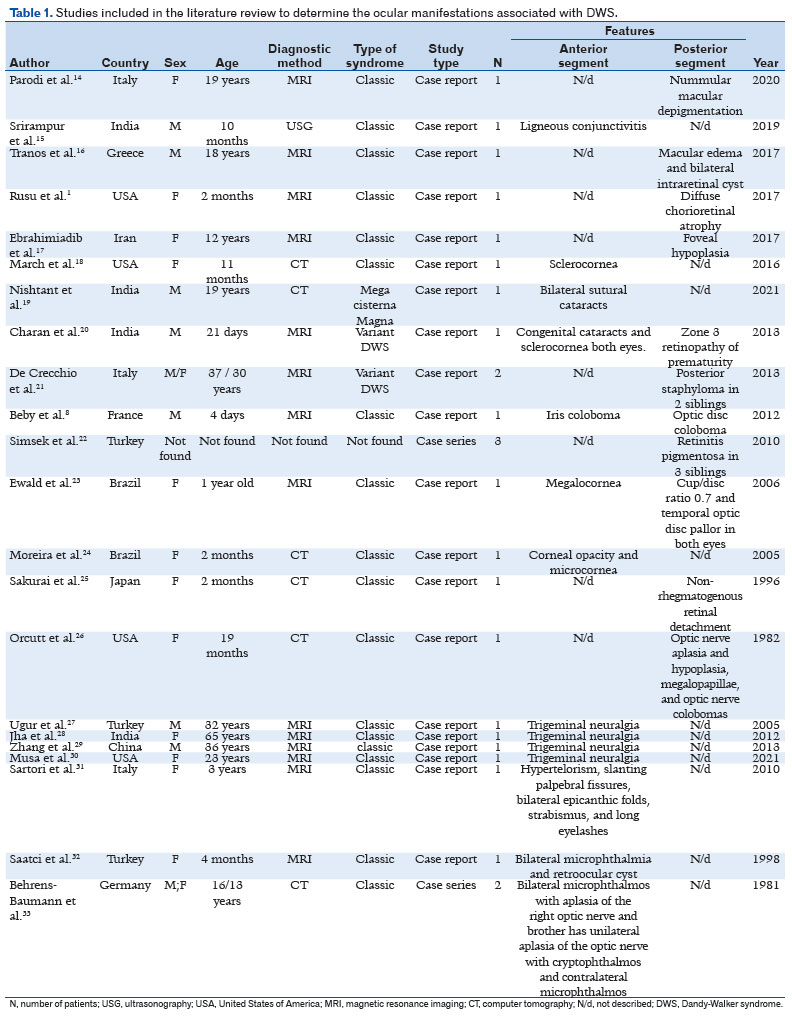
The full texts of 54 articles were read. According to the PECOT model, 22 studies were finally included. While one study was a case series of two patients, the others were all case reports. Thus, a total of 26 patients were analyzed. Of the included patients, 66% were female and 34% were male. The mean patient age was 15.6 years (SD ± 16.9 years). Approximately 57% and 42% of our sample exhibited anterior and posterior segment alterations, respectively (Table 1). Some pathologies were described according to the sequence of anterior and posterior segment structures. The following phenotypes were identified in the study patients: microphthalmia, hypotrichosis of the eyelashes and eyebrows, hypertelorism, thick eyebrows and long eyelashes, micro - and megalo-cornea, sclerocornea, bilateral cataracts, ligneous and congenital conjunctivitis, iris and optic disc coloboma, non-rhegmatogenous detachment, nummular macular depigmentation, macular edema, pathologic myopia, and trigeminal neuralgia.
DISCUSSION
The search revealed the relation of some syndromes, abnormalities of the orbit, adnexa, anterior segment, and posterior segment, and neuroophthalmological features with DWS.
Syndromic associations
Some articles have demonstrated the association between Joubert syndrome (JS) and DWS34-36. JS is an autosomal recessive disorder that is characterized by vermian hypoplasia and brainstem anomalies on imaging studies. The systemic features of JS include hypotonia, developmental delay, ataxia, irregular breathing patterns, and abnormal eye movements. The combination of additional features such as polydactyly, ocular coloboma, retinal dystrophy, renal disease, hepatic fibrosis, encephalocele, and other brain malformations define the clinical subtypes of JS. Sartori et al.37 reported a case of DWS in newborn with JS who presented with hypertelorism, slanting palpebral fissures, bilateral epicanthic folds, strabismus, and long eyelashes. DWS has also been associated with posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta, cardiac defects, and eye abnormalities syndrome (PHACE syndrome), Down's syndrome, Ritscher-Schinzel syndrome, Walker-Walburg syndrome, trisomy 18, Wisconsin syndrome, and Delleman syndrome38-41.
Orbital abnormalities
Ocular globe
Microphthalmia is one of the most common ocular congenital abnormalities. It refers to a small and disorganized ocular globe that develops due to the absence of a complete primary optic vesicle. This leads to an absent or very small eye within the orbit. The aim of microphthalmia treatment is to avoid a small orbital fossa by expanding it as early as possible.
Microphthalmia and a retrobulbar cyst were reported by Saatci et al.32 in a 4-month-old girl with an atretic parietal cephalocele and ocular malformations. It was hypothesized that these malformations had developed because the optic sac had failed to close during embryological development.
Adnexal abnormalities
Eyelashes and eyebrows
Boudghene-Stambouli et al.42 described keratitis-ichthyosis-deafness (KID) syndrome in a young girl born to non-consanguineous parents without any similar family history of hypotrichosis of the eyelashes and eyebrows. Pascual-Castroviejo et al.43 reported the case of two siblings with oculocerebrocutaneous (Delleman) syndrome, which is characterized by congenital anomalies that involve the skin, orbit, and central nervous system. One sibling exhibited a skin appendage around the eyebrow and eyeball hypoplasia. The second sibling exhibited orbital cysts and eyelid appendages. Moreira et al.24 reported hypertelorism, thick eyebrows and long eyelashes in a patient with DWS.
Anterior segment abnormalities
Cornea
Megalocornea is a rare bilateral non-progressive congenital defect that is characterized by an increased corneal diameter of more than 12.5-13 mm at birth. It results in weak zonular fibers, which can complicate cataract surgeries that require three-piece intraocular lenses23. Because the defect is anatomic, there is no cure or treatment for megalocornea. The most common interventions for megalocornea is cataract surgery, with a specific approach to individual needs, or placement of an iris-claw intraocular lens44. Ewald et al.23 reported the case of a 1-year 9-month-old female with DWS and megalocornea (right eye, 13mm; left eye, 13.5mm). The fundus examination revealed a cup/disc ratio of 0.7 and temporal optic disc pallor in both eyes. The intraocular pressure was 16mmHg in right eye and 14mmHg in the left eye. The anteroposterior diameter of both eyes was 20.55mm. However, no Haab's striae or photophobia were detected, which excluded the diagnosis of congenital glaucoma.
Microcornea is defined as a congenital abnormality in which the horizontal corneal diameter is smaller than normal. However, the globe dimensions are normal. Abnormal overgrowth of the anterior tips of the optic cup during eye development has been proposed as a cause of microcornea45. Moreira et al. and Charan et al.20,24 reported a microcornea in a 2-month-old girl and 21-day-old newborn, respectively. Other corneal abnormalities such as opacity were also observed, and computer tomography of the head was suggestive of DWS. A similar outcome related to corneal opacity has been reported by Stambolliu et al.46. They reported that 20% of patients with cataracts have DWS. March et al.18 reported sclerocornea in a 21-day-old newborn with DWS.
Lens abnormalities
Bilateral cataracts were reported by Charan et al.20 and Nishant et al.19. Furthermore, in the case report by Nishant et al. the 19-year-old male also exhibited visual field defects. The teenager had a sutural cataract, and an occipital lobe lesion was identified as the cause of the visual field defect.
Bilateral sutural cataracts is a congenital pathology of genetic origin that affects the Y-shaped sutures of the lens. The disease does not usually progress or interfere with the patient's visual acuity. For most patients, treatment is not necessary. However, surgical intervention may be required in some patients47.
Conjunctival abnormalities
Bilateral neonatal conjunctivitis has been reported by Moreira et al.24 in a 21-day-old newborn. Ligneous conjunctivitis is a rare form of chronic recurrent conjunctivitis that occurs due to plasminogen activity deficiency, which promotes fibrinous pseudomembrane formation on the tarsal and bulbar conjunctiva. Its treatment involves topical application of a fibrinolytic agent, followed by surgical excision.
Srirampur et al.15 reported ligneous conjunctivitis in a 10-month-old infant with DWS that was diagnosed on a prenatal ultrasonography at 34 weeks. The infant presented with wood-like fibrinous pseudomembranes, mainly in the right eye, without other ocular manifestations. Furthermore, hematological examination revealed a plasminogen deficiency.
Therapeutic management of conjunctivitis is long, challenging, and poorly codified. The treatment includes a combination of eye drops containing ciclosporin, heparin, and corticosteroids. Local and subconjunctival applications of fresh frozen plasma may improve the therapeutic response31.
Iris abnormalities
Congenital iris colobomas rarely require repair until the time of cataract surgery. Optic colobomas result from the failure of the optic fissure to close during embryogenesis. They can involve the iris and the structures posterior to it, up to the optic nerve. Beby et al.8 reported an iris coloboma in a 4-day-old male with bilateral posterior embryotoxon. Congenital iris colobomas vary considerably in shape, size, and location between patients. Thus, the surgical repair has to be customized to each patient's eye48.
Posterior segment
Retina
Posterior segment alterations have been reported in association with DWS. Retinal alterations in DWS include non-rhegmatogenous detachment25, nummular macular depigmentation14, and macular edema16. Non-rhegmatogenous detachment is the most common form of retinal detachment, which occurs when vitreous fluid enters through a retinal tear or hole and separates the retina from the choroid. Some retinal detachments can be treated in a clinic setting using lasers or a gas bubble injection to reattach the retina. Complex retinal detachments, characterized by multiple retinal tears or retinal scar tissue, are typically repaired via a pars plana vitrectomy or scleral buckling in an operating room49.
Optic nerve
Optic disc colobomas may occur sporadically or be inherited as an autosomal dominant inheritance. It occurs due to abnormal fusion of the two sides of the proximal end of the optic cup. These colobomas comprise a well-demarcated bowl-shaped excavation of the optic disc, which is typically decentered and deeper inferiorly50. Beby et al.8 reported an optic disc coloboma in a 4-day-old male. Ewald et al.23 reported the case of a 1-year 9-month-old female with DWS who exhibited a cup/disc ratio of 0.7 and temporal optic disc pallor in both eyes. Behrens-Baumann et al.33 reported two siblings with severe eye malformations. The 16-year-old girl had bilateral microphthalmos, aplasia of the right optic nerve, and a Dandy-Walker cyst in the cerebellum. The right eye had no optic disc or retinal vessels. Her 13-year-old brother had unilateral aplasia of the optic nerve, cryptophthalmos, and contralateral microphthalmos. Orcutt et al.26 demonstrated the relation between optic nerve aplasia and DWS.
Macula
Tranos et al.16 reported a 3-month history of reduced visual acuity (20/30) in a 18-year-old male with DWS. Fundoscopy revealed an abnormal foveal reflex in both eyes, and spectral domain optical coherence tomography revealed small intraretinal cysts in both eyes. Triamcinolone (0.05 cc of 40 mg/ml) was injected intravitreally, which was associated with some functional improvement after two months. However, the intraretinal cysts recurred after four months.
Single or multiple retinal cysts, ranging from two to ten disc diameters in size, can develop in eyes with longstanding retinal detachment. However, they rarely cause ocular symptoms or impaired central vision. Most cysts spontaneously resolve upon retinal reattachment and improved nutrition to the retina. Surgical treatment of the underlying condition often leads to complete resolution51.
Retinitis pigmentosa (RP) has also been described in relation to DWS. It is a genetically heterogeneous retinopathy caused by photoreceptor cell death and retinal pigment epithelial atrophy that eventually results in blindness in both eyes. As the disease progresses, the most effective treatment depends on the number of remaining photoreceptor cells. The treatment modalities include a neuroprotective agent, gene therapy, stem cell therapy, optogenetics, artificial retina, and chemical photoswitches52.
Simsek et al.22 reported RP in three siblings with DWS and Usher syndrome who were born to consanguineous Turkish parents. The siblings presented with RP and hearing loss. However, Usher syndrome is characterized by occasional balance problems in addition to RP and hearing loss.
Foveal hypoplasia, a retinal disorder that develops due to incomplete development of the foveal morphology, is another presentation seen in patients with DWS. Ebrahimiadib et al.17 reported non-progressive chronic visual loss in a 12-year-old female with DWS. The child had no history of refractive error or childhood ptosis or strabismus. The visual acuity was 20/50 in both eyes, and a fundus examination revealed an absent foveal reflex and mild vessel tortuosity in both eyes. Furthermore, optical coherence tomography revealed an abnormal continuity of the inner retinal layers and an absent foveal pit (grade 2 hypoplasia) in both eyes.
Posterior staphyloma (PS) is an outpouching of a circumscribed region of the posterior fundus that has been considered a hallmark of pathologic myopia. PS is characterized by a relatively abrupt scleral thinning53. De Crecchio et al.21 reported a PS in two siblings with highly myopic eyes who were diagnosed with the DWS variant. One sibling was a 37-year-old male with a history of Hodgkin's disease and trabeculectomy for high intraocular pressure 7 years prior to the PS diagnosis. The patient presented with thinning of the retinal pigment epithelium in the macular region and vitreal membrane in the peripheral retina in both eyes. The second sibling was a 30-year-old female with highly myopic eyes, a PS, and vitreal membranes in the inferior and temporal regions of the peripheral retina.
Nummular macular depigmentation is reportedly associated with DWS14. Nummular macular depigmentation is an acquired skin pigmentation disorder, which is mostly under-diagnosed. It is characterized by nummular hypopigmented lesions on the trunk of young individuals. Although several treatment options are available for this condition, topical clindamycin and benzoyl peroxide have been used traditionally. Recently, good results have been demonstrated with the use of narrow-band ultraviolet B phototherapy54. Rusu et al.1 reported retinal vascular non-perfusion in the temporal region of both eyes in 2-month-old siblings after fluorescein angiography was performed. Fundus examination revealed pink flat nerves, diffuse chorioretinal atrophy, and pigment mottling in the macula in both eyes.
Neuroophthalmological abnormalities
Ophthalmic division of the trigeminal nerve
Trigeminal neuralgia (TN) is a rare neuropathic disorder characterized by recurrent and paroxysmal episodes of short, severe, electric shock-like pain along the sensory distribution of the trigeminal nerve27. Ugur et al., Jha et al., and Zhang et al.27-29 reported TN in patients with DWS. Musa et al.30 reported the case of a 23-year-old female in whom cerebrospinal fluid build-up in a posterior fossa cyst accounted for the compression of the trigeminal nerve root, within a few millimeters of entry at the level of the pons. Medical management of TN consists of pharmacologic modalities such as antiepileptic drugs and reversible interventions such as injections of botulinum toxin type-A and local anesthetic. The surgical interventions consist of various peripheral and intracranial procedures27.
The ocular features associated with DWS have not been widely studied. The cases mentioned in this review may aid the scientific community in identifying ocular alterations in patients with DWS. However, high impact studies are required to better describe these ocular features.
REFERENCES
1. Rusu I, Gupta MP, Patel SN, Oltra E, Chan RV. Retinal vascular nonperfusion in siblings with Dandy-Walker variant. J AAPOS. 2016;20(2):174-7.
2. Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125-48.
3. Pira-Paredes SM, Montoya-Villada JH, Franco-Restrepo JL, Moncada-Velez M, Cornejo JW. [A phenotypic description of 26 patients with Ritscher-Schinzel syndrome (cranio-cerebello-cardiac dysplasia or 3C syndrome)]. Rev Neurol. 2017;64(11):481-488. Spanish.
4. Zamora EA, Das JM, Ahmad T. Dandy-Walker Malformation. [Updated 2023 Nov 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538197/
5. Delahaye A, Khung-Savatovsky S, Aboura A, Guimiot F, Drunat S, Alessandri JL, et al. 9. Am J Med Genet A. 2012;158A(10):2430-8.
6. Chi L, Zhong L, Lee D, Yu X, Caballero A, Nieman B, et al. G9a inactivation in progenitor cells with Isl1-Cre with reduced recombinase activity models aspects of Dandy-Walker complex. Biol Open. 2023;12(8):bio059894.
7. Beaman MM, Guidugli L, Hammer M, Barrows C, Gregor A, Lee S, et al. Novel association of Dandy-Walker malformation with CAPN15 variants expands the phenotype of oculogastrointestinal neurodevelopmental syndrome. Am J Med Genet A. 2023;191(11): 2757-2767.
8. Beby F, Des Portes V, Till M, Mottolese C, Denis P. Chromosome 6p25 deletion syndrome: report of a case with optic disc coloboma and review of published ophthalmic findings. Ophthalmic Genet. 2012;33(4):240-8.
9. Kotani T, Tsuda H, Ito Y, Nakamura N, Ushida T, Imai K, et al. Prenatal diagnosis of distal 13q deletion syndrome in a fetus with esophageal atresia: a case report and review of the literature. J Med Case Rep. 2022;16(1):481.
10. Santos BZ, Zancanela LB, Argemi CT, Chiquetti EMS. Terminal deletion of the short arm of chromosome 6 (6P25 3P 243): a literature review and case report of a Brazilian child/ Deleção terminal do braço curto do cromossomo 6 (6p25 3p 243): uma revisão de literatura e relato de caso de uma criança brasileira. Medicina (Ribeirao Preto, Online);55(2)abr 2022 ilus, tab. 2022.
11. Alp MY, Çebi AH, Seyhan S, Cansu A, Aydin H, Ikbal M. 22.5 MB DELETION OF 13q31.1-q34 ASSOCIATED WITH HPE, DWM, AND HSCR: A CASE REPORT AND REDEFINING THE SMALLEST DELETED REGIONS. Genet Couns. 2016;27(1):43-9.
12. Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037-42.
13. Ballarati L, Rossi E, Bonati MT, Gimelli S, Maraschio P, Finelli P, et al. 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet. 2007;44(1):e60.
14. Parodi MB, Arrigo A, Manitto MP, Bandello F. Nummular Macular Depigmentation in Dandy-Walker Syndrome. Retina. 2020;40(9): e46-e47.
15. Srirampur A, Ramappa M, Chaurasia S, Vemuganti G. Ligneous conjunctivitis in a Dandy-Walker syndrome: A rare case report. Indian J Ophthalmol. 2019;67(1):143-145.
16. Tranos P, Dervenis N, Kiouras S. Bilateral Macular Edema: A New Ocular Feature of Dandy-Walker Syndrome. Semin Ophthalmol. 2017;32(4):501-503.
17. Ebrahimiadib N, Karkheiran S, Roohipoor R, Karkhaneh R, Modjtahedi BS. Foveal hypoplasia associated with Dandy-Walker syndrome. Can J Ophthalmol. 2017;52(4):e125-e127.
18. March WF, Chalkley TH. Sclerocornea associated with Dandy-Walker cyst. Am J Ophthalmol. 1974;78(1):54-7.
19. Nishant P, Aftab N, Raj A, Jha VC, Sinha U. Adult-onset Dandy-Walker syndrome with atypical ocular manifestations. Can J Ophthalmol. 2023;58(4):e175-e177.
20. Charan GS, Narang GS, Kaur A, Kaur E. Dandy-Walker Variant Associated with Bilateral Congenital Cataract. Int J Appl Basic Med Res. 2021;11(4):277-279.
21. de Crecchio G, Cennamo G, de Leeuw N, Ventruto ML, Lonardo MC, Friso P, et al. Severe myopia with unusual retinal anomalies and Dandy-Walker sequence in two sibs. A distinct new neuro-ocular disorder. Ophthalmic Genet. 2013;34(4):254-7.
22. Simsek T, Ozdamar Y, Simsek E, Men G. Usher syndrome associated with a variant of Dandy-Walker malformation. J Pediatr Ophthalmol Strabismus. 2010;47 Online:e1-4.
23. Ewald O, Scremin F, Busch F, Von Hertwig R. [Ocular alterations in a pediatric patient with Dandy-Walker malformations: case report]. Arq Bras Oftalmol. 2006;69(1):97-9.Portuguese.
24. Moreira LMA, Neri FB, Uzeda SQ, de Carvalho AFL, Santana GC, Souza FR, Rollemberg JC. Multiple congenital malformations including severe eye anomalies and abnormal cerebellar development with Dandy-Walker malformation in a girl with partial trisomy 3q. Ophthalmic Genet. 2005;26(1):37-43.
25. Sakurai E, Shirai S, Ozeki H, Majima A. [A case of nonrhegmatogenous retinal detachment in Dandy-Walker Syndrome]. Nippon Ganka Gakkai Zasshi. 1996;100(10):832-6.Japanese.
26. Orcutt JC, Bunt AH. Anomalous optic discs in a patient with a Dandy-Walker cyst. J Clin Neuroophthalmol. 1982;2(1):43-7.
27. Ugur HC, Torun F, Yilmaz E, Kanpolat Y. Trigeminal neuralgia in a patient with Dandy-Walker malformation. J Clin Neurosci. 2005;12(7):815-7.
28. Jha VC, Kumar R, Srivastav AK, Mehrotra A, Sahu RN. A case series of 12 patients with incidental asymptomatic Dandy-Walker syndrome and management. Childs Nerv Syst. 2012;28(6):861-7.
29. Zhang W, Chen M, Zhang W. Trigeminal neuralgia due to Dandy-Walker syndrome. J Craniofac Surg. 2013;24(4):1457-9.
30. Musa J, Rahman M, Guy A, Ahmetgjekaj I, Guy A, Kola I, et al. Trigeminal neuralgia caused by Dandy-walker malformation: A case report and systematic review of the literature. Radiol Case Rep. 2021;16(10):3084-3089.
31. Sartori MT, Bartuli A, Siboni SM, Leonardi A, Valente P, Secci J, et al. Ligneous conjunctivitis and use of human plasminogen eyedrops: The Italian experience. Haemophilia. 2023;29(2):681-684.
32. Saatci I, Yelgec S, Aydin K, Akalan N. An atretic parietal cephalocele associated with multiple intracranial and eye anomalies. Neuroradiology. 1998;40(12):812-5.
33. Behrens-Baumann W, Dust G, Rittmeier K, Langenbeck U, Vogel M. [Oculo-cerebral dysplasia: aplasia of the optic nerve with familial microphthalmos and cryptophthalmus. Clinical and computer tomography study]. Klin Monbl Augenheilkd. 1981;179(2):90-3.German.
34. Buissonniere RF, Storni V, Robain O, Ponsot G. [Joubert's syndrome]. Ann Pediatr (Paris). 1990;37(3):151-6.French.
35. Cantani A, Lucenti P, Ronzani GA, Santoro C. Joubert syndrome. Review of the fifty-three cases so far published. Ann Genet. 1990; 33(2):96-8.
36. Zhang YW, Qu HB, Long N, Leng XY, Liu YQ, Yang Y. A rare mutant of OFD1 gene responsible for Joubert syndrome with significant phenotype variation. Mol Genet Genomics. 2021;296(1):33-40.
37. Sartori S, Ludwig K, Fortuna M, Marzocchi C, Calderone M, Toldo I, et al. Dandy-Walker malformation masking the molar tooth sign: an illustrative case with magnetic resonance imaging follow-up. J Child Neurol. 2010;25(11):1419-22.
38. Assari R, Ziaee V, Moghimi S, Akbari MR, Mirmohammadsadeghi A. PHACE(S) syndrome: Report of a case with new ocular and systemic manifestations. J Curr Ophthalmol. 2016;29(2):136-138.
39. Steiner JE, McCoy GN, Hess CP, Dobyns WB, Metry DW, Drolet BA, et al. Structural malformations of the brain, eye, and pituitary gland in PHACE syndrome. Am J Med Genet A. 2018;176(1):48-55.
40. Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132(3):307-11.
41. Buzenet C, Hamel-Teillac D, Acar P, Becquet F, Curan D, Michaud V, et al. [Facial hemangioma associated with arterial anomalies, coarctation of the aorta, and eye abnormalities: PHACES syndrome]. Ann Dermatol Venereol. 2000;127(3):292-5.French.
42. Boudghene-Stambouli O, Merad-Boudia A, Abdelali S. [KID syndrome, pachydermatoglyphy and Dandy-Walker syndrome]. Ann Dermatol Venereol. 1994;121(2):99-102.French.
43. Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Lapunzina P. Oculocerebrocutaneous (Delleman) syndrome: report of two cases. Neuropediatrics. 2005;36(1):50-4.
44. Sharan S, Billson FA. Anterior megalophthalmos in a family with 3 female siblings. J Cataract Refract Surg. 2005;31(7):1433-6.
45. Sun W, Xiao X, Li S, Guo X, Zhang Q. Mutational screening of six genes in Chinese patients with congenital cataract and microcornea. Mol Vis. 2011;17:1508-13.
46. Stambolliu E, Ioakeim-Ioannidou M, Kontokostas K, Dakoutrou M, Kousoulis AA. The Most Common Comorbidities in Dandy-Walker Syndrome Patients: A Systematic Review of Case Reports. J Child Neurol. 2017;32(10):886-902.
47. Moutei H, Abdellaoui M. Sutural cataract. Pan Afr Med J. 2020;36:34.
48. Ogawa GSH. Congenital iris coloboma repair with excision of colobomatous sphincter muscle. J Cataract Refract Surg. 2021;47(8):1088-1091.
49. Meier P. [Retinal detachment in children: differential diagnosis and current therapy]. Klin Monbl Augenheilkd. 2008;225(9):779-90.German.
50. Dutton GN. Congenital disorders of the optic nerve: excavations and hypoplasia. Eye (Lond). 2004;18(11):1038-48.
51. Verdaguer P, Nadal J. Intraretinal cyst secondary to longstanding retinal detachment. Eur J Ophthalmol. 2012;22(3):506-8.
52. Hristodorov D, Lohoff T, Luneborg N, Mulder GJ, Clark SJ. Investing in vision: Innovation in retinal therapeutics and the influence on venture capital investment. Prog Retin Eye Res. 2024 Mar:99:101243.
53. Zhou ZH, Xiong PP, Sun J, Wang YL, Wang JL. Effects of posterior staphyloma on choroidal structure in myopic adults: a retrospective study. BMC Ophthalmol. 2023;23(1):406.
54. Montero LC, Belinchón I, Toledo F, Betlloch I. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27(3):162-3.
AUTHORS INFORMATIONS |
|
 |
» Francisco Victor Carvalho Barroso https://orcid.org/0000-0003-3152-8839 https://lattes.cnpq.br/8207348034186978 |
 |
» Ana Laura Eloia Limão https://orcid.org/0000-0003-4661-5660 https://lattes.cnpq.br/1401230374306671 |
 |
» Manoela Gondim https://orcid.org/0000-0001-8671-2559 https://lattes.cnpq.br/5200236785788212 |
 |
» Rian Vilar Lima https://orcid.org/0000-0003-2405-1753 https://lattes.cnpq.br/6008143139000016 |
 |
» Samuel Montenegro https://orcid.org/0000-0002-4343-7769 https://lattes.cnpq.br/7322176753684670 |
 |
» João Crispim Moraes Lima Ribeiro https://orcid.org/0000-0002-8569-1159 https://lattes.cnpq.br/5238824885154872 |
Funding: No specific financial support was available for this study.
Conflict of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
February 21, 2024.
Accepted on:
June 14, 2024.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket