

João Vitor de Oliveira Pereira1,2,3; Ana Luiza Machado Ribeiro Pimentel1,2,3; Dillan Cunha Amaral2,4; Ricardo Noguera Louzada4,5; Luís Alexandre Rassi Gabriel3
DOI: 10.17545/eOftalmo/2024.0009
ABSTRACT
This report presents an exceptionally rare case of gyrate atrophy affecting the choroid and retina, accompanied by a homozygous variant of uncertain significance in the OAT gene (c.416T>G:p.M139R), which is the first documented instance in ClinVar. The case involves a 59-year-old patient who has experienced symptoms since childhood. Visual acuity was reported as light perception in the right eye and 20/100 in the left eye. Fundoscopic examination revealed characteristic chorioretinal atrophy of gyrate atrophy, along with moderate attenuation of arteriolovenular calibers. Additional diagnostic tests included ophthalmic biomicroscopy of the anterior segment, retinography/autofluorescence, Goldmann semiautomated kinetic perimetry, and dark adaptometry. Further examinations unveiled anatomical and functional abnormalities in the left eye, including multiple areas of chorioretinal gyrate atrophy, bone spicules, moderate arteriolovenular caliber attenuation, and peridiscal atrophy. The patient was subsequently referred for nutritional monitoring and prescribed a restrictive arginine diet along with vitamin B6 supplementation as part of the treatment plan.
Keywords: Gyrate atrophy; OAT; Vitamin B6; Biomarker; DNA analysis; Molecular genetics; Ophthalmic genetics; Ornithine.
RESUMO
O estudo relata um caso ultrarraro de atrofia girata de coroide e retina com variante de significado incerto homozigota no gene OAT (c.416T>G:p.M139R). O caso é de uma paciente de 59 anos com sintomas desde a infância, apresentando acuidade visual de percepção luminosa em olho direito e de 20/100 em olho esquerdo. Ao fundo de olho observou-se presença de atrofia coriorretiniana característica da atrofia girata com atenuação moderada dos calibres arteriovenulares. Foram realizados exames adicionais como biomicroscopia oftálmica de segmento anterior, retinografia/autofluorescência, perimetria visual cinética semiautomática de Goldmann e adaptometria ao escuro. Nos exames adicionais, foram registradas alterações anatômicas e disfuncionais como múltiplas áreas de atrofia girata coriorretinianas, espículas ósseas, atenuação moderada dos calibres arteriolovenulares e atrofia peridiscal no olho esquerdo. A paciente foi encaminhada para acompanhamento nutricional para tratamento com dieta restritiva de arginina e suplementação com vitamina B6.
Palavras-chave: Atrofia girata; OAT; Vitamina B6; Biomarcador; Análise de DNA; Genética molecular; Genética oftálmica; Ornitina.
INTRODUCTION
Gyrate atrophy of the choroid and retina (GACR) is a rare genetic disorder inherited in an autosomal recessive manner1. Its cause lies in a biallelic mutation occurring in the OAT gene, resulting in a deficiency of ornithine aminotransferase and subsequent hyperornithinemia2,3. Ornithine aminotransferase (OAT) is a mitochondrial enzyme responsible for converting ornithine, a derivative of arginine, into proline. Insufficient levels of this enzyme lead to the accumulation of ornithine in plasma and tissues, potentially causing cytotoxic effects on the retinal pigment epithelium (RPE)4. Symptoms commonly associated with GACR encompass nyctalopia, perimetric constriction, and decreased visual acuity. Ocular manifestations include distinct features such as multiple confluent areas of choroidal and retinal atrophy, characterized by sharply defined yellow lesions exhibiting centripetal confluence with well-defined circular borders. The term “gyrata” is derived from the Greek word gyros, meaning circle, reflecting the circular nature of these lessions5,6. Additional ocular signs comprise bone spicules, arteriolar and venular caliber attenuation, and peripapillary atrophy, often accompanied by early cataracts and myopia7. Mild sensory and motor alternations might also be present upon extraocular evaluation. The diagnosis of gyrate atrophy is typically confirmed by the detection of hyperornithinemia due to the impairment in ornithine conversion resulting from the OAT gene mutation2,8. The age of onset for this condition varies, and its exact pathophysiology remains unclear, though it is believed to commence with RPE degeneration.
In this study, we present the initial case of a patient clinically diagnosed with gyrate atrophy, associated with a variant of uncertain significance (c.416T>G) in the OAT gene.
CASE REPORT
A 59-year-old female patient visited the clinic for a follow-up GACR. She had experienced symptoms since childhood, with reported visual acuity of light perception in the right eye (OD) and 20/100 in the left eye (OS). Biomicroscopic examination revealed bilateral pseudophakia, while in the left eye, corectopia was noted due to intraocular lens synechia of the iris. Pupillary and ocular motility reflexes were found to be normal in both eyes.
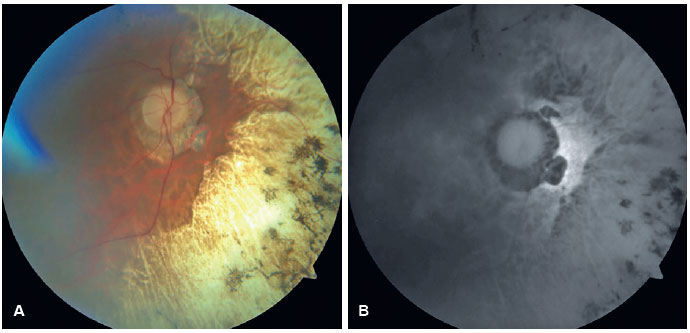
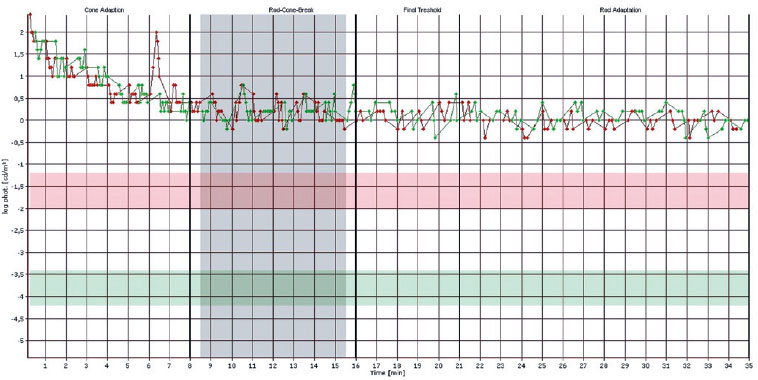
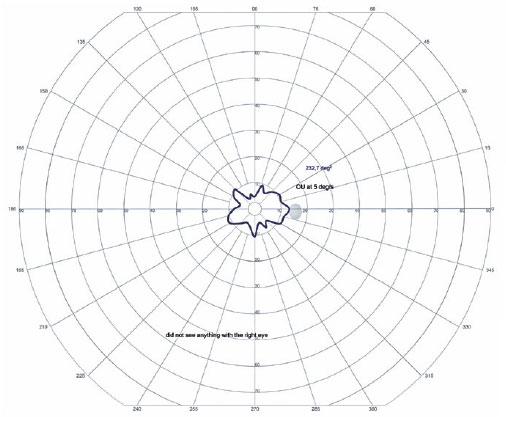
In the OD, pendular nystagmus was observed due to low vision. Fundoscopic evaluation of both eyes revealed multiple areas of chorioretinal gyrate atrophy, bone spicules, moderate attenuation of arteriolovenular calibers, and peripapillary atrophy (Figure 1A). Autofluorescence examination displayed hypoautofluorescence in the regions of atrophy (Figure 1B). Clinical assessment unveiled significant nyctalopia, as measured in the dark adaptation test, necessitating a light intensity approximately 10,000 times greater than that required by an individual with normal retinal function (Figure 2), along with perimetric constriction observed in Goldmann semiautomated kinetic perimetry (Figure 3).
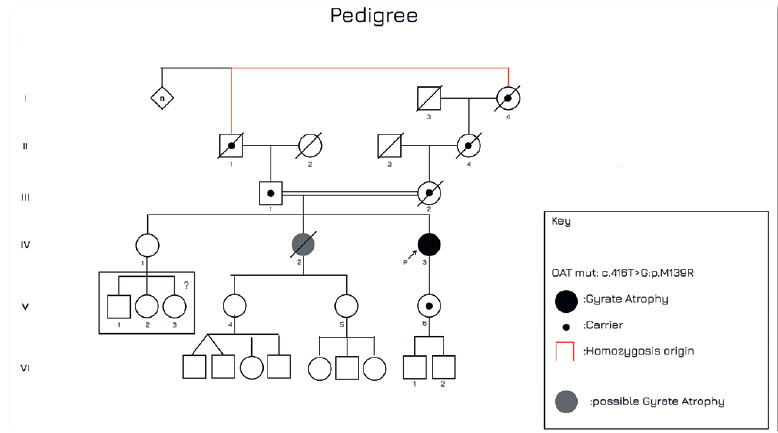
Genetic analysis revealed the presence of a homozygous variant of uncertain significance, c.416T>G:p.M139R (ClinVar:VCV0010270.8), in the OAT gene. Segregation analysis indicated that the patient's father carried the same variant in heterozygosity, as depicted in the familial pedigree (Figure 4). Bioinformatics tools including SIFT, PolyPhen-2, and Align-GVGD indicated that this genetic alteration could potentially be deleterious. Moreover, data from the gnomAD genetic database indicated a variant frequency of 0.003% among a population of over 125,000 sequenced individuals. Consequently, the patient was recommended for assessment regarding vitamin B6 and essential amino acid supplementation, alongside a prescribed restrictive arginine diet.
The genetic investigation utilized next-generation sequencing employing a commercially certified panel comprising 328 genes. Sanger sequencing was subsequently conducted to validate the identified variants. Paternal segregation analysis was performed as part of the assessment. For visual field examination, the PVCSG assessment utilized the Octopus 900 device equipped with Eye Suite software version i9.6.3.0 (Haag-Streit Diagnostics), employing a circular light projection with a diameter of 9 mm (size V), light intensity of 318 cd/m2 (4e), and a constant speed of 5º/s, with centripetal vectors in a Ganzfeld dome having 31.4 asb of luminance.
Additional diagnostic procedures, including anterior segment biomicroscopy, retinography/fundus autofluorescence, dark adaptation test, and pupillography, were conducted by the same ophthalmologist utilizing specific devices: BQ 900 biomicroscope (Haag-Streit International), Cirrus Photo 800 (Zeiss), DARKadaptometer (Roland Consult), and RETIpupillometry (Roland Consult).
DISCUSSION
This report outlines the clinical and genetic characteristics of a patient diagnosed with GACR, featuring a biallelic variant of uncertain significance, c.416T>G:p.M139R. While the patient's clinical presentation aligns with a diagnosis of gyrate atrophy, the molecular test results are not entirely conclusive.
Currently, the correlation between genotype and phenotype in GACR remains unclear1. Katagiri et al. observed that mutations in the OAT gene are associated with two distinct clinical phenotypes. Most patients exhibit gyrate atrophy, characterized by elevated plasma ornithine levels and chorioretinal degeneration. In a subset of cases, a more severe disorder manifests with neonatal-onset hyperammonemia, vomiting, and signs of encephalopathy. These patients typically demonstrate low plasma ornithine levels, indicating a primary deficiency in the reaction catalyzed by OAT during the neonatal period2.
Hyperornithinemia can stem from three recognized causes : hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, Δ(1)-pyrroline-5-carboxylate synthetase deficiency, and gyrate atrophy. Given the absence of symptoms consistent with the first two conditions, the diagnosis of gyrate atrophy is further supported by the patient's clinical presentation and genetic testing results.
While the anatomical and syndromic diagnosis of GACR in this case is typical, the identification of a variant of uncertain significance raises the question of whether it represent a novel variant. Although an identification code for this variant exists on the ClinVar website (https://www.ncbi.nlm.nih.gov/clinvar/variation/1027070/), where it was first noted in 2020 (associated with a diagnosis of retinitis pigmentosa), no published clinical data regarding this specific case are available.
The precise mechanism through which ornithine d-aminotransferase deficiency leads to chorioretinal degeneration and subsequent ornithine accumulation remains incompletely understood. The accumulation of ornithine or its metabolites can induce direct tissue damage, with the RPE being the primary site of injury in patients with GACR5. Animal model studies have indicated that sustained reduction in plasma ornithine levels can prevent retinal degeneration, while ornithine accumulation may lead to RPE degeneration7-9.
The conventional treatment for GACR typically involves prescribing vitamin B6 (pyridoxine) alongside essential amino acids and implementing an arginine-restricted diet, as arginine serves as the precursor to ornithine10. Prior research has shown that early initiation of dietary interventions restricting arginine can decelerate the progression of chorioretinal lesions. However, the efficacy of this approach remains contentious, with some studies reporting improvements or stabilization of retinal changes, while others note continued progression of chorioretinal complications. This treatment regimen necessitates meticulous monitoring, with variations in treatment initiation age, dietary adherence, and genetic heterogeneity in GACR likely contributing to the diverse outcomes10-14.
In this case, the patient underwent dietary restriction of ornithine along with vitamin B6 supplementation, which serves as a precursor to pyridoxal phosphate, a coenzyme of ornithine aminotransferase15. The slow progression of chorioretinal degenerative changes characteristics of GACR poses challenges in evaluating the efficacy of any therapeutic intervention for this condition16.
Consanguineous marriages represent a significant risk factor for the increased occurrence of genetic disorders. Hence, genetic counseling is crucial for consanguineous couples regarding family planning decisions17.
In summary, this study presents the first documented case of a patient clinically diagnosed with gyrate atrophy, associated with a homozygous variant c.416T>G (p.M139R) classified as uncertain significance in the OAT gene (ClinVar variation ID: 1027070).
REFERENCES
1. Doimo M, Desbats MA, Baldoin MC, Lenzini E, Basso G, Murphy E, et al. Functional analysis of missense mutations of OAT, causing gyrate atrophy of choroid and retina. Hum Mutat. 2013;34(1):229-36.
2. Katagiri S, Gekka T, Hayashi T, Ida H, Ohashi T, Eto Y, et al. OAT mutations and clinical features in two Japanese brothers with gyrate atrophy of the choroid and retina. Doc Ophthalmol. 2014;128(2):137-48.
3. Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina: further experience with long-term reduction of ornithine levels in children. Arch Ophthalmol. 2002;120(2):146-53.
4. Valle D, Kaiser-Kupfer MI, Del Valle LA. Gyrate atrophy of the choroid and retina: deficiency of ornithine aminotransferase in transformed lymphocytes. Proc Natl Acad Sci U S A. 1977;74(11):5159-61.
5. Takki KK, Milton RC. The natural history of gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88(4):292-301.
6. Berson EL, Schmidt SY, Shih VE. Ocular and biochemical abnormalities in gyrate atrophy of the choroid and retina. Ophthalmology. 1978;85(10):1018-27.
7. Kuwabara T, Ishikawa Y, Kaiser-Kupfer MI. Experimental model of gyrate atrophy in animals. Ophthalmology. 1981;88(4):331-5.
8. Wang T, Steel G, Milam AH, Valle D. Correction of ornithine accumulation prevents retinal degeneration in a mouse model of gyrate atrophy of the choroid and retina. Proc Natl Acad Sci U S A. 2000;97(3):1224-9.
9. Wang T, Milam AH, Steel G, Valle D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin invest. 1996;97(12):2753-62.
10. Balfoort BM, Buijs MJN, Asbroek ALMAT, Bergen AAB, Boon CJF, Ferreira EA, et al. A review of treatment modalities in gyrate atrophy of the choroid and retina (GACR). Mol Genet Metab. 2021;134(1-2):96-116.
11. Vannas-Sulonen K, Simell O, Sipilä I. Gyrate atrophy of the choroid and retina. The ocular disease progresses in juvenile patients despite normal or near normal plasma ornithine concentration. Ophthalmology. 1987;94(11):1428-33.
12. Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina: long-term reduction of ornithine slows retinal degeneration. Arch Ophthalmol. 1991;109(11):1539-48.
13. Mashima YG, Weleber RG, Kennaway NG, Inana G. Genotype-phenotype correlation of a pyridoxine-responsive form of gyrate atrophy. Ophthalmic Genet. 1999;20(4):219-24.
14. Ramesh V, McClatchey AI, Ramesh N, Benoit LA, Berson EL, Shih VE, et al. Molecular basis of ornithine aminotransferase deficiency in B-6-responsive and-nonresponsive forms of gyrate atrophy. Proc Natl Acad Sci U S A. 1988;85(11):3777-80.
15. Ohkubo Y, Ueta A, Ito T, Sumi S, Yamada M, Ozawa K, et al. Vitamin B6-responsive ornithine aminotransferase deficiency with a novel mutation G237D. Tohoku J Exp Med. 2005;205(4):335-42.
16. Santinelli R, Costagliola C, Tolone C, D'Aloia A, D'Avanzo A, Prisco F, et al. Low-protein diet and progression of retinal degeneration in gyrate atrophy of the choroid and retina: A twenty-six-year follow-up. J Inherit Metab Dis. 2004;27(2):187-96.
17. Huang J, Fu J, Fu S, Yang L, Nie K, Duan C, et al. Diagnostic value of a combination of next-generation sequencing, chorioretinal imaging and metabolic analysis: lessons from a consanguineous Chinese family with gyrate atrophy of the choroid and retina stemming from a novel OAT variant. Br J Ophthalmol. 2019; 103(3):428-35.
AUTHORS INFORMATIONS |
|
 |
» João Vitor de Oliveira Pereira https://orcid.org/0000-0001-8806-0689 http://lattes.cnpq.br/5419937219657840 |
 |
» Ana Luiza Machado Ribeiro Pimentel https://orcid.org/0000-0003-1122-6657 http://lattes.cnpq.br/8010513090617384 |
 |
» Dillan Cunha Amaral https://orcid.org/0009-0002-7948-154X https://lattes.cnpq.br/7959357721386149 |
 |
» Ricardo Noguera Louzada https://orcid.org/0000000296105768 http://lattes.cnpq.br/5978866539118374 |
 |
» Luís Alexandre Gabriel Rassi https://orcid.org/0000000204587131 http://lattes.cnpq.br/3226810848914264 |
Funding: No specific financial support was available for this study.
Conflict of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
February 27, 2023.
Accepted on:
January 21, 2024.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket