

Roberta de Julio Matheus; Julia Alves Utyama; Guilherme Novoa Colombo Barboza; Marcello Novoa Colombo Barboza; Juliana Carolina Contrera
DOI: 10.17545/eOftalmo/2021.0005
RESUMO
A Síndrome de Usher é uma doença autossômica recessiva com acometimento oftalmológico, caracterizado por Retinose Pigmentar, e otorrinolaringológico, caracterizado por surdez neurossensorial. Pode ser classificada em 3 tipos variando de acordo com o início e gravidade dos sintomas. Pacientes com essa doença apresentam perda visual progressiva, sendo o primeiro sintoma a cegueira noturna seguida de perda de campo visual periférico. Distúrbios vestibulares, como ataxia, podem estar associados. O diagnóstico da doença é importante para realização de aconselhamento genético e para iniciar reabilitação visual precoce para a melhoria da qualidade de vida do paciente.
Palavras-chave: Síndromes de Usher; Retinose pigmentar; Cegueira noturna.
ABSTRACT
Usher syndrome is an autosomal recessive disease with ophthalmological involvement, characterized by retinitis pigmentosa, and otological impairment, characterized by neurosensory deafness. It can be classified into three types, according to the time of onset and the severity of symptoms. Patients with this disease present progressive visual loss, the first symptom being nyctalopia, followed by loss of the peripheral visual field. Vestibular disorders, such as ataxia, may be associated. The diagnosis of the syndrome is important for conducting genetic counseling and for initiating early visual rehabilitation to improve the patient’s quality of life.
Keywords: Usher syndrome; Retinitis pigmentosa; Nyctalopia.
INTRODUÇÃO
A Síndrome de Usher é uma doença autossômica recessiva1 caracterizada por hipoacusia neurossensorial em graus variados e retinose pigmentar (RP)2. Apresenta incidência de 3 em cada 100 mil indivíduos na população geral2, 3 a 6% na população de deficientes auditivos2,3 e aproximadamente 50% dos casos de surdez hereditária1. Pode ser classificada em 3 tipos distintos que diferem com base no início e gravidade dos sintomas1,2. O tipo 1 é caracterizado por surdez congênita total, comprometimento vestibular e cegueira noturna na infância3,4. O tipo 2 envolve surdez congênita parcial, disfunção vestibular e cegueira noturna em adultos jovens3. O tipo 3 é caracterizado por déficit auditivo progressivo sem problemas de equilíbrio e com alterações visuais na vida adulta3.
A RP está presente em todos os tipos da doença, diferindo na idade de início2,5, sendo mais precoce no Tipo 1, que aparece na primeira década de vida5. O principal sintoma inicial notado, presente em todos os tipos, é a cegueira noturna, que lentamente se expande para alteração de visão periférica4. À fundoscopia desses pacientes é possível observar achados típicos de RP, tais como, palidez do nervo óptico, esclerose arteriolar e migração de pigmentos intrarretinianos em forma de espículas ósseas5. Também podem estar presentes: catarata subcapsular, drusa de nervo óptico, lesões foveais atróficas e edema macular cistóide, sendo este último mais comumente evidenciado com o exame de Tomografia de Coerência Óptica (OCT)5.
Exames complementares como campo visual (CV) e eletrorretinografia (ERG) podem ser úteis em casos com forte suspeita diagnóstica com ausência de sinais oftalmológicos5. No CV pode-se observar perdas de sensibilidade em diferentes fases da doença, porém é inespecífico. O ERG é o registro complexo dos potenciais elétricos da retina em resposta à estimulação luminosa, estando geralmente reduzidos ou ausentes nos pacientes com a síndrome6. Além disso pode mostrar alterações retinianas que precedem as alterações fundoscópicas, sendo o exame de escolha para o diagnóstico5.
O objetivo deste estudo foi relatar um caso de Síndrome de Usher e utilizar os achados oftalmológicos como auxílio para classificação da doença.
RELATO DO CASO
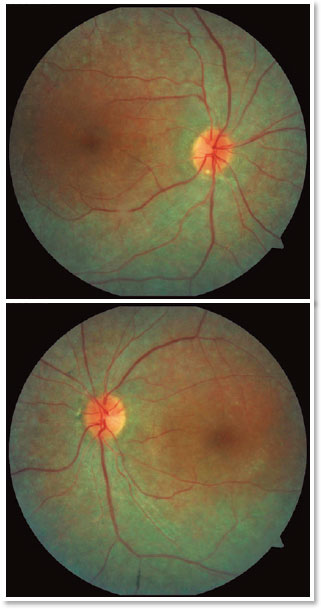
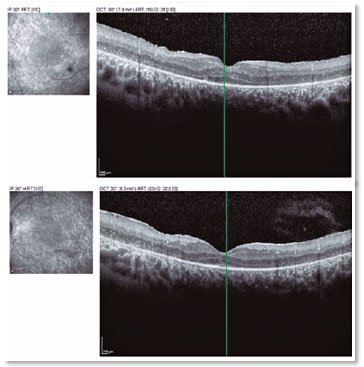
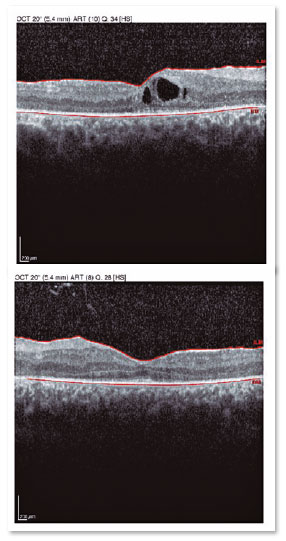
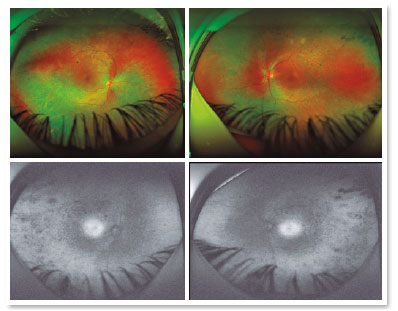
R.T.M, sexo masculino, 23 anos, branco, estudante. Queixa de baixa acuidade visual (AV) para longe e perto há 20 anos com piora progressiva há 5 anos e acometimento de visão periférica, referindo maior dificuldade à noite. Antecedentes pessoais: Atraso no DNPM, alteração de marcha - ataxia, Síndrome de Usher diagnosticada em 2007 através da ERG, surdez pré-verbal e implante coclear em 2014. Antecedentes oftalmológicos: Laudo da ERG (2007) mostrando respostas indetectáveis aos bastonetes AO e respostas reduzidas dos cones em 95% AO. Antecedentes familiares: irmã paterna com cegueira unilateral sem causa aparente. Ao exame: AV com correção: 20/200 ambos os olhos. Biomicroscopia sem alterações. PIO: OD 13mmHg e OE 14mmHg. Fundoscopia: escavação fisiológica AO. À Retinografia: nervo corado, escavação fisiológica, vasos preservados, mobilização do epitélio pigmentar da retina, múltiplos pontos hipopigmentados difusamente distribuídos preservando a mácula AO (Figura 1). O exame de OCT mostrou irregularidade na zona elipsóide com preservação na região foveal, presença de estrutura hiperrefletiva acima da Membrana Limitante Interna (MLI) configurando uma membrana epirretiniana e redução na espessura da camada nuclear externa parafoveal AO (Figura 2). Presença de cavidades císticas hiporrefletivas intrarretinianas evidenciando edema de mácula em OD (Figura 3). Ao Optomap observou-se áreas de hiperpigmentação na periferia, as espículas ósseas AO (Figura 4). Paciente passou por avaliação do setor da retina que indicou injeção de Dexametasona intravítrea (Ozurdex), porém devido às suas restrições de mobilidade, consequentes da síndrome, não retornou ao serviço, o que impossibilitou avaliar sua evolução clínica com o uso da medicação.
DISCUSSÃO
A Síndrome de Usher é caracterizada por hipoacusia neurossensorial associada a perda visual progressiva, tendo como principais sintomas visuais a cegueira noturna e perda de campo visual periférico inicialmente. Ao analisar o caso, pode-se afirmar que o paciente acima apresenta Síndrome de Usher tipo 1, devido às alterações visuais no início da vida adulta de caráter progressivo associada à surdez pré-verbal e alteração na marcha, como descrito.
O correto diagnóstico é importante, uma vez que medidas para melhorar a qualidade de vida e o aconselhamento genético podem ser realizadas o quanto antes2,6. Diagnosticar a doença apenas através de achados clínicos pode ser um desafio, sendo necessária a análise da genética molecular2 e exames complementares, como ERG, OCT e Retinografia com autofluorescência. Até hoje, onze genes associados a doença já foram identificados7.
A ERG, exame que avalia a resposta dos fotorreceptores à estímulos luminosos através de eletrodos em contato com a córnea, é de grande importância para avaliação da perda da função dessas estruturas e pode trazer informações sobre o prognóstico e resposta ao tratamento8. O achado mais relevante, em pacientes com a Síndrome é a diminuição ou ausência de resposta de cones e bastonetes6. Essa característica se deve ao acometimento frequente e severo da camada nuclear externa da retina, que contém os núcleos dos fotorreceptores, em paciente com RP9. Já a camada nuclear interna da retina composta por células amácrinas, bipolares e horizontais, normalmente estão preservadas nessa doença, porém também podem apresentar danos de acordo com sua evolução9. Na maioria dos casos, a perda de função dos bastonetes é superior à dos cones9, como observado no paciente relatado. Vale ressaltar que muitas vezes esse achado na ERG antecede as alterações fundoscópicas8 e clínicas, pois pode haver uma perda de 90% da função dos cones e ainda assim o paciente apresentar uma boa acuidade visual, dessa forma, exames objetivos, como a ERG são melhores para o diagnóstico precoce9.
O exame de OCT é não invasivo e útil para avaliação da morfologia da retina, particularmente da mácula, sendo capaz de mensurar a espessura retiniana, avaliar a qualidade da camada de fotorreceptores, além de determinar a presença de EMC9 e membrana epirretiniana (MER)3. Tanto a presença de EMC quanto de MER na Síndrome de Usher, ocorre devido ao processo inflamatório presente nesta entidade, que pode alterar a permeabilidade do Epitélio Pigmentar da Retina (EPR), levando a esses achados. Ao exame observa-se presença lesões hiporrefletivas intrarretinianas compatíveis com líquido e espessamento foveal, além de membrana hiperrefletiva acima da MLI, caracterizando EMC e MER respectivamente8. Por avaliar o status da camada de fotorreceptores, OCT é fundamental para predizer o quanto o tratamento pode impactar na evolução da doença e consequentemente no prognóstico desses pacientes9.
A Retinografia com autofluorescência é útil para documentar a deterioração do EPR, que se caracteriza por acúmulo de lipofuscina local, iniciando-se na periferia da retina8,9. Áreas hiperautofluorescentes no exame são aquelas que produzem as menores amplitudes na ERG, sendo assim, são as que apresentam maior dano do EPR local9, podendo ser observadas com frequência nesses pacientes.
O manejo oftalmológico da Síndrome é atenuar e evitar a progressão da RP melhorando a qualidade de vida. Atualmente, há na literatura muitos estudos em andamento sobre novas estratégias terapêuticas como o implante artificial de retina10,11, terapia farmacológica12,13 e terapia gênica14.
Os implantes artificiais de retina consistem em microchips, que entram em contato com a camada fotorreceptora15-17, ou eletrodos implantados na parte interna da retina em contato com as células ganglionares, de tal forma que uma melhora significativa da capacidade de leitura e reconhecimento de objetos já foi relatada nesses casos18. Existem também estudos em andamento que avaliam a possibilidade de transplante de EPR, fotorreceptores19 ou células-tronco20, com resultados promissores, como aumento da AV21 e proteção dos neurônios retinianos22.
Sobre a abordagem com terapia farmacológica existem evidencias de suplementação com Vitamina A, e nesses pacientes houve uma perda mais lenta de campo visual9, porém deve-se manter um acompanhamento periódico para avaliação de possíveis efeitos colaterais, tais como osteoporose9. O ácido docosa-hexaenoico, derivado do ômega-3, também tem sido utilizado para tratamento, uma vez que as rodopsinas e iodopsinas contêm níveis elevados dessa substância e pacientes com RP apresentam níveis séricos mais baixos9.
Para tratamento do EMC, injeções intravítreas de esteróides ou anti VEGF, como Bevacizumabe, podem ser indicados8, além de Inibidores da anidrase carbônica, orais ou tópicos, que podem trazer uma melhora transitória da AV nesses pacientes23-25. Medidas que melhorem a AV como a extração da catarata e redução da exposição à luz também podem ser benéficas a esses pacientes9. Em casos mais avançados, auxílios ópticos para visão subnormal podem melhorar a qualidade de vida do paciente, tais como dispositivos eletrônicos de ampliação como computador portátil e tablets26,27.
Em relação à terapia gênica foi evidenciado uma restauração significativa da visão em pacientes jovens próximos à cegueira8. Porém maiores estudos devem ser feitos para melhoria da biocompatibilidade e estabilidade desses tratamentos a longo prazo18,26,28.
Ao avaliar o caso relatado, foi possível observar o padrão clínico característico da Síndrome: paciente adulto jovem, sintomas iniciais de RP associado à surdez neurossensorial e alterações de marcha. Muitos pacientes acabam abandonando o tratamento devido às dificuldades de mobilidade e a pouca melhora observada com os tratamentos disponíveis. Porém apesar do caráter progressivo e do prognóstico visual reservado da doença, o avanço tecnológico dos tratamentos disponíveis tem se mostrado capazes de melhorar a qualidade de vida dos pacientes. Sendo assim é de extrema importância encorajá-los a manter o acompanhamento oftalmológico e otorrinolaringológico regular.
REFERÊNCIAS
1. Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25(1):42-9.
2. Benson, MD, MacDonald M. Bilateral uveitis and Usher syndrome: a case report. J Med Case Rep. 2015 Mar 15;9:60.
3. Liarth JCS, Gonçalves EA, Gonçalves JOR, Neiva DM, Leal FAM. Síndrome de Usher: características clínicas. Arq Bras Oftalmol. 2002;65(4):457-61.
4. Testa F, Melillo P, Rossi S, Marcelli V, Benedicts A, Colucci R, et al. Prevalence of macular abnormalities assessed by optical coherence tomography in patients with Usher syndrome. Ophtalmic Genetics. 2017;39(1):17-21.
5. Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher Syndrome: Hearing Loss with Vision Loss. Adv Otorhinolaryngol. 2011;70:56-65.
6. Mendieta L, Berezovsky A, Salomão SR, Sacai PY, Pereira JM, Fantini SC. Acuidade visual e eletrorretinografia de campo total em pacientes com síndrome de Usher. Arq Bras Oftalmol. 2005; 68(2):171-6.
7. Jouret G, Poirsier C, Spodenkiewicz M, Jaquin C, Gouy E, Arndt C, et al. Genetics os Usher Syndrome: New Insights From a Meta-analysis. Otol Neurotol. 2019;40(1):121-9.
8. Buchaim G, Rezende MP, Maia M. Implante intravítreo de liberação crônica de dexametasona (Ozurdex®) para o tratamento de edema macular por retinose pigmentar: relato de caso. Arq Bras Oftalmol. 2013;76(6):377-9.
9. Hartong DT, Berson EL, Dryja TP. Retinitis Pigmentosa. Lancet. 2006 Nov 18;368(9549):1795-809.
10. Djilas M, Oles C, Lorach H, Bendali A, Dégardin J, Dubus E, et al. Three-dimensional electrode arrays for retinal prostheses: modeling, geometry optimization and experimental validation. J Neural Eng. 2011;8(4):046020.
11. Wilke R, Gabel VP, Sachs H, Schmidt KUB, Gekeler F, Besch D, et al. Spatial resolution and perception of patterns mediated by a subretinal 16-electrode array in patients blinded by hereditary retinal dystrophies. Invest Ophthalmol Vis Sci. 2011;52(8):5995- 6003.
12. Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher syndrome. Arch Ophthalmol. 2010;128(9):1146-50.
13. Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther. 2006;6(7):717-26.
14. Zou J, Luo L, Shen Z, Chiodo VA, Ambati BK, Hauswirth W, et al. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest Ophthalmol Vis Sci. 2011;52(5):2343-51.
15. Zrenner E. Will retinal implants restore vision? Science. 2002; 295(5557):1022-5.
16. DeMarco PJ Jr, Yarbrough GL, Yee CW, McLean GY, Sagdullaev BT, Ball SL, et al. Stimulation via a subretinally placed prosthetic elicits central activity and induces a trophic effect on visual responses. Invest Ophthalmol Vis Sci. 2007;48(2):916-26.
17. Zrenner E, Bartz-Schmidt KU, Benav H, Besch D, Bruckmann A, Gabel VP, et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc Biol Sci. 2011; 278(1711):1489-97.
18. Berger AS, Tezel TH, Del Priore LV, Kaplan HJ. Photoreceptor transplantation in retinitis pigmentosa: short-term follow-up. Ophthalmology. 2003;110(2):383-91.
19. Seiler MJ, Aramant RB. Transplantation of neuroblastic progenitor cells as a sheet preserves and restores retinal function. Semin Ophthalmol. 2005;20(1):31-42.
20. Radtke ND, Aramant RB, Seiler MJ, Petry HM, Pidwell D. Vision change after sheet transplant of fetal retina with retinal pigment epithelium to a patient with retinitis pigmentosa. Arch Ophthalmol. 2004;122(8):1159-65.
21. Meyer JS, Katz ML, Maruniak JA, Kirk MD. Embryonic stem cell-derived neural progenitors incorporate into degenerating retina and enhance survival of host photoreceptors. Stem Cells. 2006;24(2):274-83.
22. Fishman GA, Gilbert LD, Fiscella RG, Kimura AE, Jampol LM. Acetazolamide for treatment of chronic macular edema in retinitis pigmentosa. Arch Ophthalmol. 1989;107(10):1445-52.
23. Chen JC, Fitzke FW, Bird AC. Long-term eff ect of acetazolamide in a patient with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1990;31(9):1914-8.
24. Reis RF, Moreiro-Gonçalves N, Silva SE, Brandão EM, Falcão-Reis FM. Comparison of Topical Dorzolamide and Ketorolac Treatment for Cystoide Macular Edema in Retinitis Pigmentosa and Usher’s Syndrome. Ophthalmologica. 2015;233(1):43-50.
25. Berson EL, Rabin AR, Mehaff ey L III. Advances in night vision technology: a pocketscope for patients with retinitis pigmentosa. Arch Ophthalmol. 1973;90(6):427-31.
26. Stieglitz T. Development of a micromachined epiretinal vision prosthesis. J Neural Eng. 2009;6(6):065005.
27. Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329(5990):413-7.
INFORMAÇÃO DOS AUTORES




Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
24 de Janeiro de 2020.
Aceito em:
17 de Agosto de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket