

Heitor A. Simões1; Luiz Arthur F. Beniz2; Vitor B. P. Pereira1; Jonas E. N. Franco Neto 3; Arnaldo P. Cialdini1; José Beniz Neto5
DOI: 10.17545/eOftalmo/2020.0019
ABSTRACT
Here, we report a case of acute posterior multifocal placoid pigment epitheliopathy (APMPPE) that was treated satisfactorily with oral and topical corticosteroids, along with topical non-hormonal antiinflammatory drugs, with complete visual recovery. The patient presented with significant visual impairment and multiple perimacular yellowish-white placoid lesions; and evaluation of multimodal imaging revealed significant changes and the patient responded suitably to the proposed therapy. APMPPE is an idiopathic retinochoroidopathy that causes a sharp, acute, and painless visual loss and often leads to visual sequelae that may even affect the central nervous system. Better visual prognosis can be ensured by differentiating this disease from white dots syndrome and administering treatment in the early stages of the disease. However, there is no well-established treatment for APMPPE thus far.
Keywords: Posterior uveitis; Chorioretinitis; Choroiditis; Retina
RESUMO
Relato de um caso de Epiteliopatia Pigmentar Placóide Multifocal Posterior Aguda (EPPMPA) que foi tratada satisfatoriamente com corticoesteroide oral e tópico, associados a anti-inflamatório não hormonal tópico, com recuperação visual completa. A paciente se apresentou com baixa visual importante, múltiplas lesões placoides branco-amareladas perimaculares e alterações significativas na avaliação de imagem multimodal, obtendo excelente resposta a terapia proposta. A EPPMPA é uma retinocoroidopatia idiopática que causa perda visual acentuada, aguda e indolor e frequentemente cursa com sequelas visuais que podem até acometer o sistema nervoso central. A diferenciação da síndrome dos pontos brancos e o início do tratamento precoce resultam em um melhor prognostico visual. Seu tratamento ainda não está bem estabelecido.
Palavras-chave: Uveíte posterior; Coriorretinite; Coroidite; Retina.
INTRODUCTION
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE), first described by Gass in 1968, is an inflammatory, idiopathic, and self-limited ophthalmological condition characterized by the acute onset of multiple yellowish-white placoid lesions in the posterior pole region and mid-peripheral retina 1. It is included in a group of idiopathic retinochoroidopathies called “white dot syndromes 2.” This condition is often preceded by flu-like symptoms such as fever, headache, or malaise, and may subsequently even affect the central nervous system 3. The etiology of APMPPE remains unknown, but its association with viral infections and vaccines suggests that it may be an autoimmune process 4. Initially, it was assumed that this disorder mainly affected the retinal pigment epithelium (RPE) 5. However, subsequent evidences suggested that it primarily occurred in the choroid 6. Typically, APMPPE affects healthy individuals, regardless of their gender, aged 20-30 years, particularly in myopics 2. It is characterized by photopsia, followed by sudden and painless visual loss with central and paracentral scotomas. The disease is usually bilateral but can also be asymmetric. Visual acuity may be normal or severely impaired depending on the location of the lesions 3,7. The prognosis is generally favorable, with excellent visual acuity recovery from weeks to months, although most patients may have some visual sequelae 2,8,9.
CASE REPORT
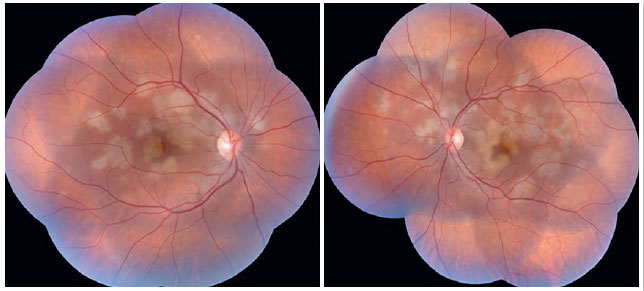
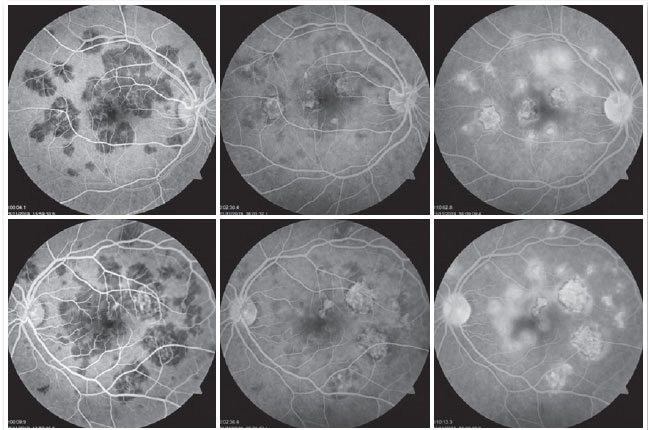
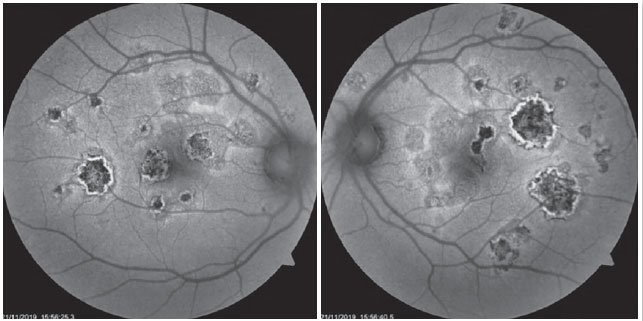
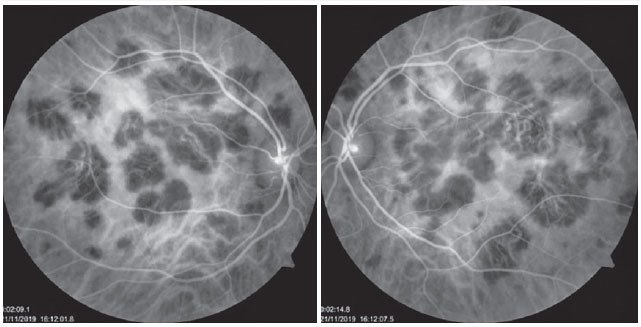
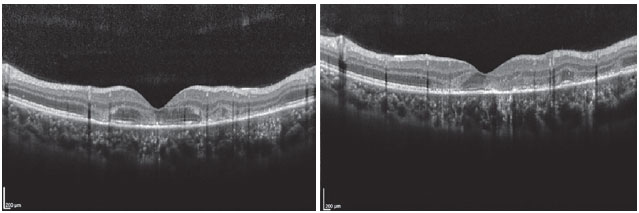
The patient was a 27-year-old female civil engineer from Araguaína-TO. She sought care at the Centro Brasileiro de Cirurgia de Olhos (CBCO) on 11/21/2019, complaining of spots in the visual field bilaterally, particularly worse in the left eye, that had appeared one week prior. She did not have a family history of any ocular or systemic diseases. During examination, she presented with best corrected visual acuity (BCVA) in the right eye (OD) 20/100 (-0.50 -0.25 × 170º) and left eye (OS) 20/80 (-0.25 -0.25 × 170º), anterior segment without changes and the presence of trace cell in the anterior vitreous of both eyes (OU). Funduscopic examination revealed yellowish-white plaques in the perimacular and nasal regions (Figure 1). Intraocular pressure was 15mmHg (OD) and 14mmHg (OS). In the multimodal analysis of images, the fluorescein angiography (FA) presented with lesions with early hypofluorescence and late staining (Figure 2), autofluorescence (AF) showed 3 hypoautofluorescent lesions in O.U. that suggested chronicity, and 5 lesions in the O.D. and 4 lesions in the hyperautofluorescent O.S. that indicated acute injuries (Figure 3). Finally, indocyanine green (ICG) showed hypocyanescent lesions (Figure 4). Optical coherence tomography (OCT) showed slight neurosensory retinal detachment, with the presence of the inflammatory material below the internal retina and involvement of the ellipsoid zone (Figure 5). The patient also underwent electroretinogram (ERG) and electroculogram (EOG), both without alterations. She was consequently diagnosed with APMPPE, and treatment with topical ketorolac tromethamine every 6 hours, fluormetholone acetate every 2 hours, and prednisone 60mg orally, with weekly regressions, for 6 weeks was initiated. On 02/20/2020, her VA recovered to 1.0 in both eyes along with an improvement in the funduscopic aspects of the lesions (Figure 6).

DISCUSSION
APMPPE is a disease that may be preceded by flu-like symptoms, such as fever, headache, or malaise 3. In addition, it may affect the central nervous system, and may lead to the occurrence of diffuse headaches or complications such as cerebral vasculitis or meningoencephalitis 10. Burés-Jestrup A. et al reported 3 patients in a series of 16 cases who had nonspecific viral symptoms 11. Our patient only presented with a mild headache.
Funduscopic findings compatible with the disease are characterized by multifocal placoid yellowish-white lesions, with varying dimensions and located in the posterior pole and mid-periphery. This characteristic aspect was well visualized during the funduscopic examination of our patient. In addition, these lesions gradually evolve into areas of hypopigmentation and pigment accumulation due to atrophy and focal proliferation of RPE 12.
The use of complementary imaging tests can help in disease diagnosis and treatment indication. In fluorescent angiography (FAG), the lesions presented with early hypofluorescence due to internal choroid perfusion abnormalities with late hyperfluorescence (early blockade, with late staining). RPE atrophy may concurrently produce window defect hyperfluorescence 10. Furthermore, we noted two previously reported conditions in this case report. In the case series reported by Burés-Jestrup A. et al., all the patients presented with identical pattern characteristics of APMPPE 11. In AF, hyperautofluorescence is observed in areas with active inflammation that, with healing, has the hypoautofluorescent aspect 13. The first outcome is observed when the patient arrives at our service, and second when the condition is resolved and the patient shows visual recovery. ICG (indocyanine green) is superior to FAG in showing capillary perfusion failures, and the acute lesions appear hypocyanescent. Additionally, it shows gradual improvement as the disease is eventually treated 14.
OCT shows heterogeneous accumulation of subretinal fluid in the early stages of the disease, evolving to the development of hyperreflectivity in the external retina, caused by a rupture process in the inner segment layer and external segments, along with RPE hyperreflectivity that can last for up to three months 15,16. We found that in the reports of Xerri et al., 3 of the 20 eyes had subretinal fluid and 7 eyes had alterations in the external segments of the retina 17.
By observing the images of this case report, it is possible to conclude that this is a classic case of APMPPE. This diagnosis is based mainly on clinical and funduscopic examination, and is complemented by the abovementioned imaging exams. Differential diagnoses include evanescent white dot syndrome, multifocal choroiditis and panuveitis, internal puncttata choroidopathy, serpiginous choroiditis, Birdshot retinochoroidopathy, acute retinal pigment epithelitis, multifocal retinal metastases, and intraocular lymphoma 7.
The indication for treatment for patients with APMPPE remains in debate. We treated our patient with a combination of topical and oral corticosteroids, along with topical non-steroidal anti-inflammatory drugs, owing to the fact that some authors recommend indicating treatment in the presence of macular or central nervous system involvement. In these cases, oral corticosteroids are preferred at the initial dosage of 1mg/kg, following which the dosage can be regressed within a few weeks 11,16,18,19.
The patient presented in this case report showed a particularly characteristic condition of APMPPE. The clinical evolution was favorable and there was a complete recovery of the VA, which was similar to the outcomes of other similar reports.
REFERENCES
1. Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80(2):177-85.
2. Mirza RG, Jampol LM. White spot syndromes and related diseases. In: Ryan SJ, ed. Retina. Philadelphia, PA: Saunders; 2013:1337-1381.
3. Jones NP. Acute posterior multifocal placoid pigment epitheliopathy. Br J Ophthalmol. 1995;79(4):384-9.
4.Deutman AF, Oosterhuis JA, Boen-Tan TN, Aan de Kerk AL. Acute posterior multifocal placoid pigment epitheliopathy. Pigment epitheliopathy of choriocapillaritis? Br J Ophthalmol. 1972;56(12):863-74.
5. Spaide RF. Autofluorescence imaging of acute posterior multifocal placoid pigment epitheliopathy. Retina. 2006;26(4):479-82.
6. Dhaliwal RS, Maguire AM, Flower RW, Arribas NP. Acute posterior multifocal placoid pigment epitheliopathy. An indocyanin green angiographic study. Retina. 1993;13(4):317-25.
7. Quillen DA, Davis JB, Gottlieb JL, Blodi BA, Callanan DG, Chang TS, et al. The white dot syndromes. Am J Ophthalmol. 2004;137(3):538-50.
8. Fiore T, Iaccheri B, Androudi S, Papadaki TG, Anzaar F, Brazitikos P, et al. Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina 2009;29(7):994-1001.
9. Wolf MD, Alward WL, Folk JC. Long-term visual function in acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1991;109(6):800-3.
10. Souka AA, Hillenkamp J, Gora F, Gabel V-P, Framme C. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol 2006;244(10):1219-23
11. Bures-Jelstrup A, Adan A, Casaroli-Marano R. Acute posterior multifocal placoid pigment epitheliopathy. Study of 16 cases. Arch Soc Esp Oftalmol. 2007;82(5):291-7.
12. Park D, Schatz H, McDonald HR, Johnson. Indocyanine green angiography of acute multifocal posterior placoid pigment epitheliopathy. Ophthalmology. 1995;102(12):1877-83.
13. Goldenberg D, Habot-Wilner Z, Loewenstein A, Goldstein M. Spectral domain optical coherence tomography classification of acute posterior multifocal placoid pigment epitheliopathy. Retina. 2012;32(7):1403-10.
14. Birnbaum AD, Blair MP, Tessler HH, Goldstein DA. Subretinal fluid in acute posterior multifocal placoid pigment epitheliopathy. Retina. 2010;30(5):810-4.
15. Fiore T, Iaccheri B, Androudi S, Papadaki TG, Anzaar F, Brazitikos P, et al. Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina 2009;29(7):994-1001.
16. Wolf MD, Alward WL, Folk JC. Long-term visual function in acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1991;109(6):800-803.
17. Xerri O, Salah S, Monnet D, Brézin AP. Untreated Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): a case series. BMC Ophthalmol. 2018 Mar 20;18(1):76.
18. Grkovic D, Oros A, Bedov T, Karadzic J, Gvozdenovic L, Jovanovic S. Acute posterior multifocal placoid pigment epitheliopathy-retinal “white dot syndrome”. Med Glas (Zenica). 2013;10(1):194-6.
19. Thomas BC, Jacobi C, Korporal M, Becker MD, Wildemann B, Mackensen F. Ocular outcome and frequency of neurological manifestations in patients with acute posterior multifocal placoid pigment epitheliopathy (APMPPE). J Ophthalmic Inflamm Infect. 2012;2(3):125-31.
AUTHOR’S INFORMATION





Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
August 27, 2020.
Accepted on:
October 13, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket