

Natália Maia de Faria1; Lívia Freire Reis2; Josué Geraldo Lessa3
DOI: 10.17545/eoftalmo/2018.0036
RESUMO
INTRODUÇÃO: A síndrome de Bardet-Biedl é uma doença de herança autossômica recessiva rara cujas principais manifestações clínicas são polidactilia, obesidade, retardo mental, hipogenitalismo e distrofia retiniana que causa progressiva perda visual a partir da infância.
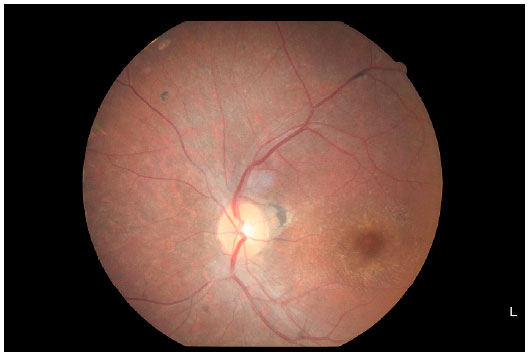
RELATO DE CASO: Paciente, 17 anos, sexo masculino, apresentou baixa acuidade visual progressiva em ambos os olhos. Apresentava déficit intelectual e má formação cardíaca desde o nascimento, assim como estrabismo divergente. Além disso, tinha baixa estatura, obesidade e polidactilia de mãos e pés. Ao exame oftalmológico, constatou-se acuidade visual com correção de 0,1 pela tabela de Snellen em ambos os olhos, pressão intraocular e biomicroscopia normais. À fundoscopia, observou-se descoloração do epitélio pigmentado da retina dando ao fundo uma coloração acinzentada e um aspecto mosqueado, diminuição do calibre vascular, presença de palidez de papila, migração de pigmento em padrão de espículas ósseas, inclusive macular, e aumento do reflexo foveal.
CONCLUSÃO: A distrofia retiniana está presente em cerca de 90% dos casos já descritos na síndrome de Bardet-Biedl e muitas vezes é o que caracteriza o diagnóstico. Tal fato denota a importância do oftalmologista tanto na investigação inicial quanto no seguimento de afecções sistêmicas.
Palavras-chave: Distrofias Retinianas; Retinose Pigmentar; Síndrome de Bardet-Biedl.
ABSTRACT
INTRODUCTION: Bardet-Biedl syndrome is a rare autosomal recessive disease with the chief clinical manifestations of polydactylism; central obesity; mental retardation; hypogenitalism; and retinal dystrophy, which leads to progressive visual loss since childhood.
CASE REPORT: A 17-year-old boy presented with progressive low visual acuity in both eyes. He had cardiac malformation since birth, intellectual disability, divergent strabismus, short stature, obesity, and polydactyly of hands and feet. Ophthalmologic examination using Snellen chart revealed a visual acuity of 0.1 in both eyes and normal intraocular pressure and biomicroscopy result. Fundoscopy revealed discoloration of the retinal pigment epithelium, giving the fundus a grayish color and mottled appearance; a decrease in vessel diameter; paleness of the optic papilla; deposits of macular and peripheral bone spicule-shaped pigments; and increase in foveal reflex.
CONCLUSION: Retinal dystrophy, which occurs in approximately 90% of patients with Bardet-Biedl syndrome, usually confirms its diagnosis. This manifestation indicates the importance of the role of an ophthalmologist in the initial investigation and follow-up of systemic complications.
Keywords: Retinal Dystrophies; Retinitis Pigmentosa; Bardet-Biedl Syndrome.
RESUMEN
INTRODUCCIÓN: El síndrome de Bardet-Biedl es una enfermedad de herencia autosómica recesiva rara cuyas principales manifestaciones clínicas son polidactilia, obesidad, retardo mental, hipogonadismo y distrofia retiniana que causa progresiva pérdida visual desde la infancia.
REPORTE DE CASO: Paciente, 17 años, sexo masculino, presentó baja acuidad visual progresiva en ambos ojos. Presentaba déficit intelectual y malformación cardíaca desde el nacimiento, así como estrabismo divergente. Además, tenía baja estatura, obesidad y polidactilia en manos y pies. Al examen oftalmológico, se constató acuidad visual con corrección de 0,1 por la tabla de Snellen en ambos ojos, presión intraocular y biomicroscopía normales. Al realizarse la fundoscopía, se ha observado decoloración del epitelio pigmentado de la retina, lo que le daba al fondo una coloración agrisada y un aspecto moteado, disminución del calibre vascular, presencia de palidez papilar, emigración de pigmento en calidad de espículas óseas, incluso macular, y aumento del reflejo foveal.
CONCLUSIÓN: la distrofia retiniana está presente en cerca del 90% de los casos ya descritos en el síndrome de Bardet-Biedl y muchas veces es lo que caracteriza el diagnóstico. Tal hecho denota la importancia del oftalmólogo tanto en la averiguación inicial como en el seguimiento de afecciones sistémicas.
Palabras-clave: Distrofias Retinianas; Retinitis Pigmentosa; Síndrome de Bardet-Biedl.
INTRODUÇÃO
A síndrome de Bardet-Biedl (SBB) é uma doença de herança autossômica recessiva rara com uma prevalência que varia de 1:160.000 a 1:13.5001. As principais manifestações clínicas são distrofia retiniana, polidactilia, obesidade, retardo mental e hipogenitalismo. A distrofia retiniana é o achado mais consistente e causa progressiva perda visual a partir da infância2. Neste presente trabalho é descrito um caso com achados compatíveis com o diagnóstico de síndrome de Bardet-Biedl.
RELATO DO CASO
O paciente D.C.S, 17 anos, masculino, branco, estudante, natural de, e residente em, Belo Horizonte, foi encaminhado ao Instituto de Olhos Ciências Médicas devido história de baixa acuidade visual progressiva em ambos os olhos. Segundo a acompanhante, o paciente apresentava déficit intelectual e má formação cardíaca desde o nascimento, assim como estrabismo (Figura 1) e uso de tampão na infância.
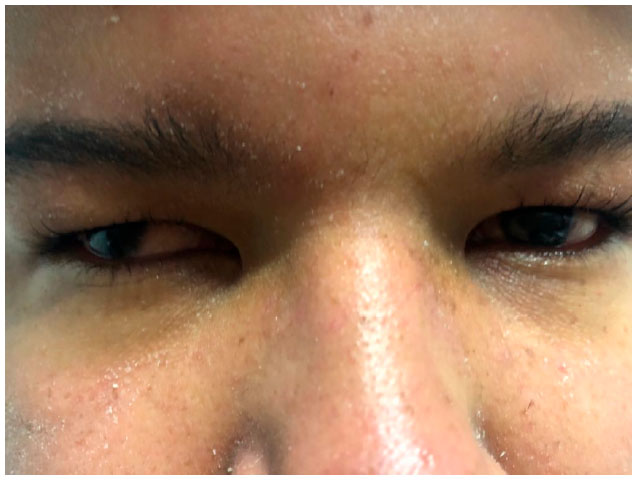
As provas de função renal, sódio e potássio séricos, glicemia, testosterona, hormônios folículos-estimulantes (FSH) e luteinizante (LH), apresentaram, todos, resultados normais. A história familiar mostrava pais não consangüíneos e uma prima materna falecida por problema cardíaco não informado.
À ectoscopia, apresentava baixa estatura, obesidade e polidactilia de mãos e pés (Figuras 2 e 3).
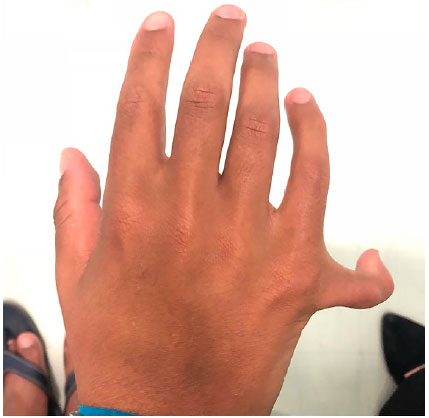
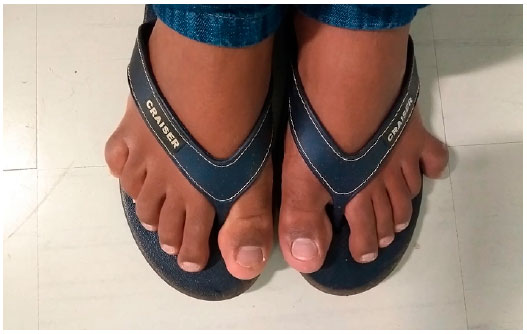
Ao exame oftalmológico, constatou-se acuidade visual com correção de 0,1 pela tabela de Snellen em ambos os olhos. A pressão intraocular e a biomicroscopia eram normais. À fundoscopia, observou-se palidez de papila, alteração do epitélio pigmentar retiniano e redução do calibre vascular de ambos os olhos (AO). O exame de campo visual mostrou-se não confiável em AO.
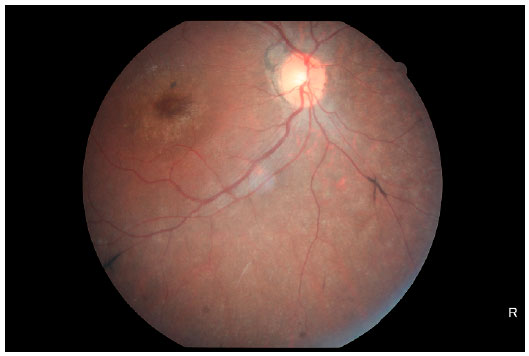
Na retinografia (Figuras 4 e 5) observamos características importantes desta distrofia tapetorretiniana, tais como descoloração do epitélio pigmentado da retina (EPR) dando ao fundo uma coloração acinzentada e um aspecto mosqueado, diminuição do calibre vascular, mobilização de pigmento em periferia e macular com padrão de espículas ósseas e aumento do reflexo foveal.
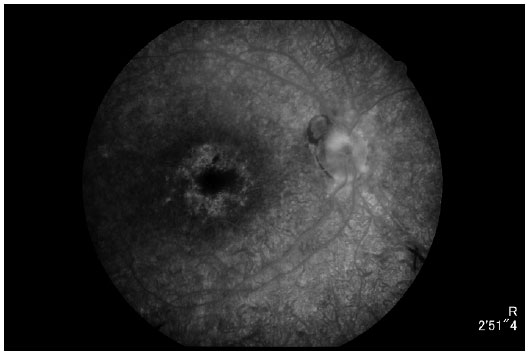
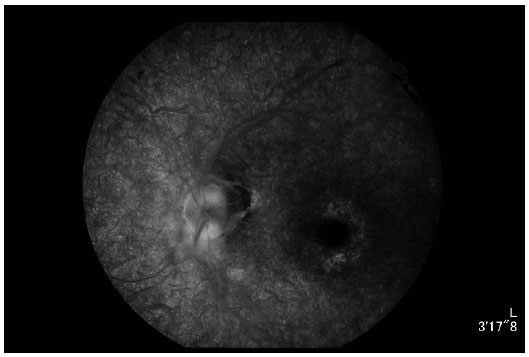
À angiografia (Figuras 6 e 7) destaca-se presença de hiperfluorescência transmitida difusamente, sugestiva de rarefação do EPR; hipofluorescência por bloqueio tanto peridiscal quanto perivascular, sugestiva de mobilização de pigmento; e hiperfluorescência por defeito em janela sugestiva de atrofia do EPR perifoveal.
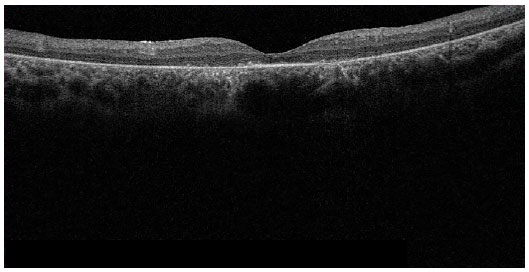
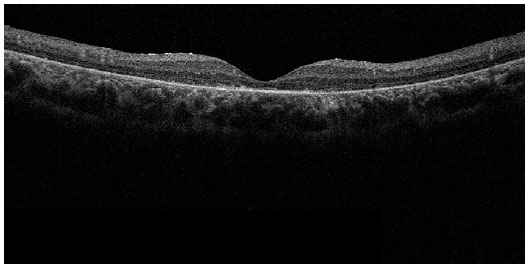
À tomografia de coerência óptica (figuras 8 e 9), observa-se afinamento difuso da retina, principalmente às custas de retina externa, apresentando perda difusa de fotorreceptores.
DISCUSSÃO
A SBB é um distúrbio genético autossômico recessivo, configurada como uma ciliopatia, em que os genes associados à doença estão associados à função ciliar primária3.
Apresenta extensa heterogeneidade genética e pelo menos 20 genes já foram mapeados em diferentes cromossomos2.
As principais características clínicas são a distrofia de cone-bastonete, com perda de visão na infância acompanhada por cegueira noturna; polidactilia pós-axial; obesidade troncular; deficiência intelectual; hipogenitalismo masculino e más formações geniturinárias femininas complexas; e disfunção renal, que é uma das principais causas de morbidade e mortalidade3.
As características secundárias incluem anomalias oculares como estrabismo, catarata e astigmatismo, distúrbios da fala ou atrasos, braquidactilia ou sindactilia, atrasos no desenvolvimento, ataxia, diabetes mellitus, dismorfismo craniofacial, diabetes insípido nefrogênico, fibrose hepática e cardiopatia congênita. Beales7 e colaboradores sugeriram que a presença de quatro características primárias ou três primárias mais duas secundárias é diagnóstica. As raras associações incluem hipotireoidismo, doença de Hirschsprung, epilepsia, anomalias genitais, estenose anal e uma dentição anormal2.
O estudo genético molecular por testes clinicamente disponíveis pode ser usado para confirmar o diagnóstico. As correlações genótipo-fenótipo não são claras; assim, não há estratégia recomendada para testes com um único gene. Neste caso, é prudente sequenciar os genes que são mais comumente mutados2.
Devido à lenta emergência e variabilidade da expressão de suas características clínicas, o diagnóstico de BBS em uma determinada pessoa pode ser retardado1. Além disso, existem outras síndromes, além da de Bardet-Biedl, que apresentam combinações de achados como defeitos oculares, retardo mental, hipoplasia genital, obesidade e anomalias digitais. Embora menos frequentes, as síndromes de Laurence-Moon, Alstrom e Biemond II devem ser incluídas no diagnóstico diferencial2.
O desenvolvimento de distrofia retiniana é a alteração mais comum que guia a investigação clínica e leva ao diagnóstico2,4. As funções, tanto dos bastonetes como a dos cones, são afetadas, culminando em uma apresentação atípica da retinose pigmentar com envolvimento macular precoce. A maculopatia pode estar associada, ou não, à degeneração da retina periférica 5,6.
Os primeiros sinais de disfunção da retina podem não ser aparentes até a idade de sete a oito anos, quando a cegueira noturna ocorre de maneira insidiosa7. Pela segunda a terceira década de vida, a mácula está envolvida em todas as pessoas e é acompanhada por acuidade visual de 0,1 ou pior. Em um estudo anterior, 63,6% dos indivíduos afetados eram legalmente cegos aos 20 anos de idade8.
Campos visuais são geralmente anormais aos dez anos de idade e evoluem com uma perda anual reportada de até três graus por ano. Normalmente, pouco mais de uma ilha central de visão permanece aos, aproximadamente, 20 anos de idade1.
Na maioria dos casos, a eletrorretinografia (ERG) é anormal e tipicamente possui respostas muito reduzidas ou extintas, com altos limiares de adaptação ao escuro. A degeneração retiniana progressiva e a baixa confiabilidade do teste de campo visual nesta síndrome são responsáveis pela falta de correlação entre a máxima resposta do ERG e as áreas residuais do campo visual. ERGs podem ser anormais até os 14 meses de idade, mas distrofia significativa de cone-bastonete não é aparente na maioria das crianças menores de cinco anos e a cooperação com o teste de ERG nessa idade é freqüentemente ruim. A menos que seja fortemente indicado, o teste de ERG pode ser adiado até pelo menos quatro anos de idade2.
Outros achados oftalmológicos podem incluir nistagmo, estrabismo, alta miopia, catarata e glaucoma2.
A polidactilia pós-axial está presente em mais de 90% dos casos, e pode ser apenas um achado dismórfico único ao nascimento, eventualmente acometendo os quatros membros, o lado ulnar das mãos ou o lado fibular dos pés, isoladamente. Tal achado pode ser verificado juntamente com braquidactilia e/ou sindactilia parcial (comumente entre o segundo e o terceiro dedos do pé)5.
Obesidade está presente em grau moderado em cerca de 90% dos pacientes, assim como no caso relatado1. Aparece na infância e permanece problemática durante toda a vida adulta e deve ser gerenciada com dieta, exercício e terapias comportamentais.
O hipogenitalismo é freqüentemente presente em homens devido, provavelmente, a um distúrbio primário da síndrome genética4.
Anormalidades do metabolismo da glicose como intolerância aos carboidratos e diabete mellitus dos tipo I e II, têm sido descritas entre os afetados. Portanto, pacientes portadores de síndrome de Bardet-Biedl devem ser rastreados para detectar a presença de alterações no metabolismo da glicose.
A insuficiência renal é a principal causa de morbidade e de óbito entre os pacientes com síndrome de Bardet-Biedl. Portanto, é importante um monitoramento da função renal do paciente2.
Em resumo, a SBB pode ser detectada com base em manifestações clínicas sem a necessidade de recursos laboratoriais. Nefrologistas podem ajudar a diagnosticar a SBB entre pacientes com insuficiência renal. Os casos de SBB em unidades de diálise podem servir como índices para o rastreamento da síndrome e, ao fazê-lo, fornecer mais dados sobre a prevalência de SBB em países como o Brasil, onde não há dados sobre a incidência da síndrome.
O aconselhamento genético é imprescindível para pacientes e seus familiares, devido à herança autossômica recessiva da doença2. Há relatos de pacientes do sexo feminino que procriaram apesar da alta taxa de infertilidade na SBB, tornando importantes as orientações de planejamento familiar2,4.
CONCLUSÃO
As características clínicas da SBB primárias incluem distrofia retiniana, polidactilia pós-axial, obesidade central, comprometimento cognitivo, hipogonadismo masculino, más formações geniturinárias femininas complexas e disfunção renal.
O caso descrito apresenta três critérios maiores (polidactilia, dificuldade de aprendizagem, distrofia retiniana) e pelo menos dois menores (cardiopatia, atraso de desenvolvimento, estrabismo), concluindo o diagnóstico presumível para a síndrome relatada. A distrofia retiniana está presente em cerca de 90% dos casos já descritos e muitas vezes é o que caracteriza o diagnóstico. Tal fato denota a importância do oftalmologista tanto na investigação inicial quanto no seguimento de afecções sistêmicas.
REFERÊNCIAS
1. Lavinsky J, Goldhardt R, Ariente SK, Domingues CG, Lavinsky F. Síndrome de Bardet-Biedl: relato de dois casos. Arq Bras Oftalmol. 2003; 66(5). Acessado em: 28/10/2018. Disponível em: http://www.scielo.br/pdf/%0D/abo/v66n5/18160.pdf.
2. Forsythe E, Beales PL. Bardet-Biedl syndrome. 2003 Jul (Updated 2015 Apr 23). In: Adam MP, Ardinger HH, Pagon RA, et al. (ed.). GeneReviews® (Internet). Seattle: University of Washington; 1993-2018. Acessado em: 28/10/2018. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1363/.
3. Mockela A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Progress in Retinal and Eye Research. 2011; 30(4). Acessado em: 28/10/2018. Disponível em: https://www.sciencedirect.com/science/article/pii/S1350946211000164.
4. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Human Gen. 2013; 21. Acessado em: 28/10/2018. Disponível em: https://www.nature.com/articles/ejhg2012115.
Financiamento: declaram não haver
Parecer CEP: não se aplica
Conflitos de interesse: Declaram não haver
Recebido em:
8 de Outubro de 2018.
Aceito em:
7 de Novembro de 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket