

Arthur Borges dos Santos1; Antônio Bandeira e Silva2; Mateus Abdalla Bastos3; Daniel de Almeida Wanderley Guedes4; Kátia Delalibera Pacheco5; Fabrício Tadeu Borges6; Marcos Pereira de Ávila7
DOI: 10.17545/eoftalmo/2018.0035
RESUMO
As distrofias em padrão do epitélio pigmentar da retina constituem um grupo heterogêneo de doenças hereditárias da mácula que têm origem em mutações do gene periferina/RDS. São caracterizadas pelo acúmulo de lipofucsina no epitélio pigmentar da retina que resultam em diferentes padrões fenotípicos e podem ser confundidas com drusas. Em alguns casos o curso não é favorável pois há formação de atrofia geográfica ou neovascularização de coroide. Apresentamos um caso de distrofia padrão do epitélio pigmentar da retina em um paciente do sexo masculino com baixa visão associada a descolamento de epitélio pigmentar da retina e tubulação da retina externa em fóvea do olho esquerdo cujo principal diagnóstico diferencial foi realizado com a degeneração macular relacionada à idade.
Palavras-chave: Distrofias Retinianas; Epitélio Pigmentado da Retina; Degeneração Macular.
ABSTRACT
Retinal pigment epithelium pattern dystrophies comprise a heterogeneous group of hereditary diseases of the macula that originate from the mutations of the peripherin/RDS gene. They are characterized by the accumulation of lipofuscin in the retinal pigment epithelium, which results in different phenotypical patterns and may be confused with drusen. In certain cases, the course is not favorable, due to geographic atrophy formation or choroidal neovascularization. Here, we report a case of pattern dystrophy of the retinal pigment epithelium in a male patient with poor vision, associated with retinal pigment epithelium detachment and external retinal tubulation in the fovea of the left eye. The main differential diagnosis was age-related macular degeneration.
Keywords: Retinal Dystrophies; Retinal Pigment Epithelium; Macular Degeneration.
RESUMEN
Las distrofias en calidad del epitelio pigmentario de la retina constituyen un grupo heterogéneo de enfermedades hereditarias de la mácula que tienen origen en mutaciones del gene periferina/RDS. Se caracterizan por el acúmulo de lipofucsina en el epitelio pigmentario de la retina que resultan en diferentes estándares fenotípicos y se pueden confundir con drusas. En algunos casos, el curso no es favorable, pues hay formación de atrofia geográfica o neovascularización coroidea. Presentamos un caso de distrofia estándar del epitelio pigmentario de la retina en un paciente del sexo masculino con baja visión asociada al desprendimiento de epitelio pigmentario de la retina y tubulación de la retina externa en fóvea del ojo izquierdo, cuyo principal diagnóstico diferencial ha sido realizado con la degeneración macular relacionada a la edad.
Palabras-clave: Distrofias Retinianas; Epitelio Pigmentado de la Retina; Degeneración Macular.
INTRODUÇÃO
Distrofias em padrão do epitélio pigmentar da retina constituem um grupo de doenças com herança autossômica dominante, associadas a mutações no gene periferina/RDS, que apresentam acúmulo de lipofucsina ao nível do epitélio pigmentado da retina (EPR) ou no espaço sub-retiniano1,2.
A disposição de acúmulo dos pigmentos define os fenótipos clínicos e frequentemente mais de uma apresentação pode coexistir3. A apresentação clínica possui semelhanças com outras doenças que atingem a mácula como a degeneração macular relacionada à idade (DMRI), doença de Stargardt e coriorretinopatia serosa central (CRSC)2,4.
Os pacientes com distrofia padrão podem permanecer assintomáticos ao longo da vida ou apresentarem sintomas visuais que variam desde um discreto embaçamento visual e metamorfopsia até a perda da visão central por formação de atrofia geográfica ou neovascularização de coroide4.
Apresentamos um caso de distrofia padrão cujo principal diagnóstico diferencial foi realizado com a DMRI.
RELATO DE CASO
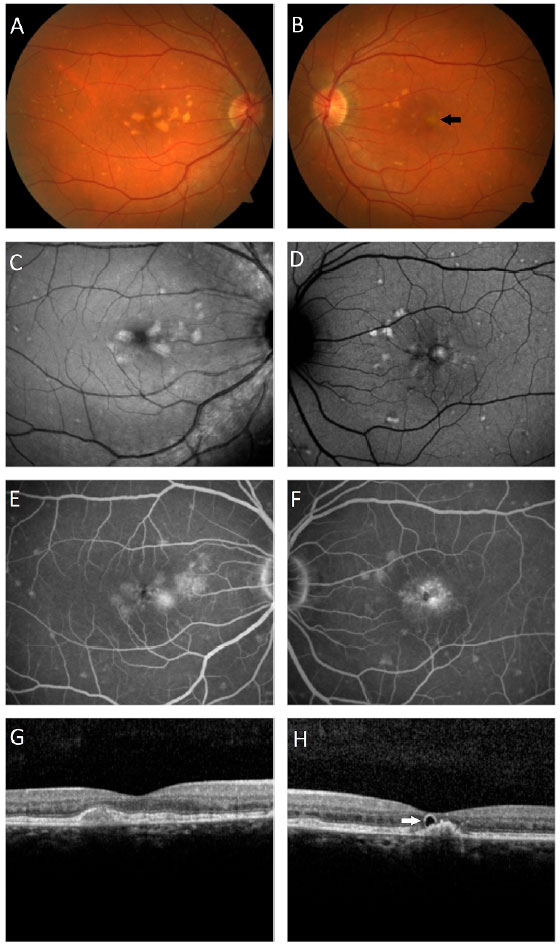
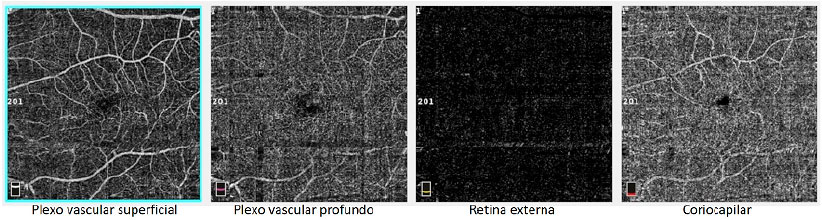
Paciente do sexo masculino, branco, 46 anos de idade, apresentou queixa de embaçamento visual progressivo em olho esquerdo com início há 6 meses, porém com melhora parcial espontânea observada nos últimos 2 meses. Ao exame oftalmológico apresentava acuidade visual com correção de 20/20 em olho direito e 20/60 em olho esquerdo. Não possuía alterações no exame biomicroscópico e a pressão intraocular era de 14mmHg em ambos os olhos. À fundoscopia em ambos os olhos foram observados depósitos sub-retinianos de coloração amarelada (flecks-like) distribuídos em polo posterior e média periferia com aparente confluência em região central da mácula (Figura 1A, B). Na fóvea do olho esquerdo visualizava-se lesão arredondada, elevada e com hipopigmentação do EPR (Figura 1B). O exame de autofluorescência evidenciou múltiplas áreas hiperautofluorescentes, correspondentes aos locais dos depósitos (Figura 1C, D). A fluoresceinografia apresentou áreas difusas de hiperfluorescência tardia por impregnação com maior intensidade em região macular em ambos os olhos (Figura 1E, F). A tomografia de coerência óptica spectral-domain (OCT-SD) em olho direito mostrou a presença de material hiperrefletivo entre a retina neurossensorial e o complexo EPR-membrana de Bruch (figura 1G), enquanto em olho esquerdo foi observado descolamento do epitélio pigmentar da retina (DEP) e tubulação da retina externa em região foveal (figura 1H). Complementou-se a investigação diagnóstica com exame de angiografia por OCT pela possibilidade de existir neovascularização de coroide em local do DEP em olho esquerdo, porém não foram evidenciados sinais de fluxo no angiograma (Figura 2). O Eletrorretinograma de campo total mostrou disfunção retiniana difusa de cones e bastonetes em ambos os olhos e o eletro-oculograma apresentava-se sub-normal. Não foi realizado teste genético para avaliar mutação do gene periferina/RDS. Foi orientado ao paciente realizar acompanhamento clínico regular e testes com grade de Amsler semanalmente.
DISCUSSÃO
As distrofias em padrão são um grupo heterogêneo de doenças maculares, geralmente diagnosticadas em torno da 2ª ou 3ª década de vida, que podem apresentar semelhanças com outras afecções que atingem a mácula, dentre elas, a DMRI é uma das causas mais frequentes de confusão diagnóstica e tratamentos desnecessários nesses pacientes1,2,5. Isso ocorre pela aparência dos depósitos sub-retinianos que muitas vezes são confundidos com drusas e a possibilidade de desenvolver áreas de hipopigmentação/atrofia do EPR ou neovascularização de coroide em pacientes com idade mais avançada6. Em estudo realizado por Ozkaya et al. apenas 45% dos pacientes obtiveram o diagnóstico correto de distrofia padrão e os outros 55% foram diagnosticados como DMRI, CRSC ou alterações inespecíficas do EPR2.
Os achados clínicos que distinguem as distrofias em padrão da DMRI incluem a idade precoce de aparecimento da doença, a ausência de drusas em seu formato característico e a presença de pigmentos amarelo-acinzentados em vários padrões na área macular7. Esse material composto por lipofucsina tem origem na disfunção fagocitária dos segmentos externos dos fotorreceptores pelo EPR determinada pela mutação no gene periferina/RDS localizado no cromossomo6,4,8.
Na distrofia padrão o exame de autofluorescência mostra áreas de hiperautofluorescência nos locais de acúmulo do pigmento e hipoautofluorescência em áreas atróficas do EPR9. A fluoresceinografia é útil para investigação de neovascularização de coroide e para diferenciar a distrofia padrão da doença de Stargardt pois não há silêncio coroideo3,10. No exame de OCT pode ser identificada desorganização citoarquitetural das camadas externas da retina, espessamento do EPR e material hiperrefletivo, correspondentes aos depósitos de lipofucsina, localizados ao nível do EPR ou entre a retina neurossensorial e o complexo EPR-membrana de Bruch2,11. Os exames de eletro-oculograma e eletrorretinograma, principalmente o multifocal, podem estar alterados6,12,13.
Embora a maioria dos casos de distrofia em padrão apresentem bom prognóstico visual, a perda da visão central em idades avançadas pode acontecer em até 50% dos casos, sendo proporcionada pela formação de atrofia geográfica ou neovascularização de coroide8,13,14. Se houver formação de membrana neovascular o tratamento envolve o uso de agentes anti-VEGF isoladamente ou concomitante com PDT, porém há relatos de casos que evidenciaram remissão espontânea da neovascularização sem necessidade de tratamento6,7,8,13.
No caso descrito, o paciente apresentou achados característicos de distrofia padrão multifocal simulando fundus flavimaculatus em ambos os olhos, que é caracterizada pela presença dos depósitos amarelados (flecks-like) em polo posterior e além das arcadas vasculares4,15. Em olho direito foi possível visualizar também o subtipo em forma de asas de borboleta pela presença dos depósitos dispondo-se em projeções em formato de asas a partir do centro foveal1,16. Ainda na fundoscopia, o aspecto dos depósitos na região macular semelhantes a drusas moles confluentes em ambos os olhos, a hipopigmentação foveal com elevação do EPR em olho esquerdo e a presença de tubulação da retina externa evidenciada no exame de OCT desse olho sugeriram inicialmente o diagnóstico de DMRI. Essa hipótese diagnóstica foi afastada pois o paciente apresentava idade inferior à compatível com a doença, as lesões em média periferia diferiam do aspecto clínico das drusas e no OCT-SD os depósitos estavam localizados predominantemente no espaço sub-retiniano. O relato do paciente em apresentar baixa visão em olho esquerdo, com melhora parcial do sintoma nos últimos meses, juntamente com a análise dos achados do OCT neste olho em região foveal, sugeriam a presença de neovascularização de coroide no local, porém foi descartada esta hipótese após exame de angiografia por OCT. A formação do DEP, portanto, pode estar relacionada à presença de material viteliforme sub-EPR17, enquanto a presença de tubulação da retina externa representa consequência do dano prévio ocasionado pela lipofucsina aos fotorreceptores, com posterior reabsorção do depósito18.
Este relato acrescenta à literatura um caso de distrofia padrão do epitélio pigmentar da retina, com apresentação de mais de um subtipo da doença, cujo principal diagnóstico diferencial foi com a DMRI. Verificouse ainda no OCT-SD a presença de tubulação da retina externa em região foveal do olho esquerdo, um achado pouco descrito na literatura em casos de distrofia padrão, porém com mais relatos na DMRI. Conclui-se que diante da suspeita de distrofia padrão do epitélio pigmentar da retina faz-se necessário utilizar as ferramentas diagnósticas disponíveis para se evitar a confusão diagnóstica com outras doenças de maior prevalência e os tratamentos desnecessários.
REFERÊNCIAS
1. Isaac DLC, Santos RAV, Ávila M. Distrofia em forma-de-borboleta: relato de caso. Arq Bras Oftalmol. 2007; 70(1):129-32.
2. Ozkaya A, et al. Clinical and imaging findings of pattern dystrophy subtypes; Diagnostic errors and unnecessary treatment in clinical practice. J Fr Ophtalmol. 2018; 41(1):21-9.
3. Houly JR. Pattern dystrophy of retinal pigment epithelium. Eye Care Vis. 2017; 1(1):1-2.
4. Silva R, Farah ME. Manual de retina. Lisboa: Lidel Edições Técnicas; 2015.
5. Marmor MF, McNamara A. Pattern dystrophy of the retinal pigment epithelium and geographic atrophy of the macula. Am J Ophthalmol. 1996; 122(3):382- 92.
6. Parodi MB, Pozzo S, Ravalico G. Photodynamic therapy for choroidal neovascularization associated with pattern dystrophy. Retina. 2003; 23(2):171-6.
7. Anastasakis A, et al. Spontaneous regression of choroidal neovascularization in a patient with pattern dystrophy. Case Rep in Ophthalmol Med. [online journal]. 2016 [Acessado em: 02/06/2018]; 2016: 9685290 [about 5p.]. Disponível em: https://www.hindawi.com/journals/criopm/2016/9685290/.
8. Empeslidis T, Vardarinos A, Deane J, Banerjee S. Intravitreal ranibizumab in the treatment of butterfly-shaped pattern dystrophy associated with choroidal nevascularization: a case report. Case Rep Ophthalmol. 2012; 3(1):77-82.
9. Boon CJF, Klevering BJ, Keunen JEE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008; 48(26):2569-77.
10. Boon CJF, Schooneveld MJ, Hollander A, Lith-Verhoeven JJC, Zonneveld-Vrieling MN, Theelen T, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007; 91(11): 1504-11.
11. Hannan SR, Salvo G, Stinghe A, Shawkat F, Lotery AJ. Common spectral domain OCT and electrophysiological findings in diferent pattern dystrophies. Br J Ophthalmol. 2013; 97(5):605-10.
12. Duinkerke-Eerola KU, Pinckers A, Cruysberg JRM. Pattern dystrophy of the retinal pigment epithelium. Int Ophthalmol. 1987; 11(2):65-72.
13. Parodi MB, Iacono P, Cascavilla M, Zucchiatti I, Kontadakis DS, Bandello F. Intravitreal bevacizumab for subfoveal choroidal neovascularization associated with pattern dystrophy. Invest Ophthalmol Vis Sci. 2010; 51(9):4358-61.
14. Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol. 2005; 89(9):1115-9.
15. Roy R, Kumar S, Chandrasekharan, Ghose A, Sharma P. Multimodal imaging in multifocal pattern dystrophy simulating fundus flavimaculatus. Indian J Ophthalmol. 2016; 64(5):395-6.
16. Saatci AO, Yasti ZO, Köse S, Memişoglu B. Butterfly-like pattern dystrophy and unilateral choroidal neovascularization. Acta Ophthalmol Scand. 1998; 76(6):734-6.
17. Grob S, Yonekawa Y, Eliott D. Multimodal imaging of adult-onset foveomacular vitelliform dystrophy. Saudi J Ophthalmol. 2014; 28(2):104-10. 18. Goldberg NR, Greenberg JP, Laud K, Tsang S, Freund KB. Outer Retinal Tubulation in degenerative retinal disorders. Retina. 2013; 33(9):1871-6.







Financiamento: declaram não haver
Parecer CEP: não se aplica
Conflitos de interesse: Declaram não haver
Recebido em:
21 de Agosto de 2018.
Aceito em:
6 de Novembro de 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket